
Abstract
The radioactivity concentrations for 90Sr and 137Cs in soil samples collected near Fukushima Daiichi nuclear power station were investigated. The depth profile of 137Cs from the surface soil to 20 cm showed a typical decreasing tendency, that is, high radioactivity from the surface down to 5 cm due to the strong sorption of specific minerals. After deposition of 90Sr, 90Sr has migrated to deeper soil layers in the past 5 years compared to 137Cs. This tendency was supported by the results of sequential extraction to identify the predominant sorption species, and by the sorption coefficients determined by batch-wise sorption experiments.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The severe accident at Fukushima Daiichi nuclear power station (FDNPS) on 11 March 2011 was caused by the tsunami that followed the great earthquake of magnitude 9.0. Hydrogen explosions, a breach of reactor pressure vessels, and vent operations occurred in reactors, resulting in the release into the atmosphere of enormous amounts of radionuclides contained within the fuel. Highly radioactive plumes, containing fission products such as 134Cs, 135Cs, 137Cs, 131I, 129mTe, and 90Sr, were transported from the reactor buildings to the local and regional environment [1,2,3]. Several years after the accident, many researchers have been involved in tracking and monitoring hazardous radionuclides, especially the gamma emitter 137Cs with a half-life of ca. 30 years [4]. Both wet and dry deposits of radioactive Cs have been investigated [5, 6]. Sanada et al. [7] measured the distribution of air dose rates and the distribution of radioactive cesium deposition on the ground within a radius of approximately 5 km from the NPS, using an air-borne survey system. The footprint of several radioactive plumes with quite high gamma radioactivity that extended from the FDNPS was illustrated. Meanwhile, less emission of Sr has been estimated and this attributed to its lower volatility. In a report by the Ministry of Education, Culture, Sports, Science and Technology (MEXT) [8], it is noted that a short half-life radionuclide (89Sr), which is a characteristic product of nuclear accidents, was detected in 5 cm-thick soil from the surface collected from a wide area of Fukushima prefecture, while a significant but quite low contamination (over 10 Bq kg−1 dry soil) of 90Sr, with a half-life of around 30 years, was observed in limited samples collected from the NPS, mainly along the north-west direction and at very few isolated points. A similar study conducted by the government and Fukushima prefecture also showed significant contamination in some locations in Fukushima [9]. Near the main gate of FDNPS in 2011, a soil sample with a high 137Cs activity concentration of 1,790,000 Bq kg−1 contained a 90Sr concentration of only 1070 Bq kg−1 [10]. In a recent study by Rosenberg et al. [11], the depth profiles of 137Cs and 90Sr in soil collected at the Fukushima exclusion zone were investigated. They found the high activity level of Cs and the tendency of a near exponential decline with depth down to 15 cm. On the other hand, 90Sr concentrations at most 385 Bq kg−1 showed 3–4 orders lower than 137Cs. Due to the low radioactivity of 90Sr, only two soil layers at 0–2.5 cm and 2.5–5 cm were analyzed.
In such early studies after the accident, an evaluation of the transport ratio was attempted. This is the ratio of the activity concentration of 90Sr to that of 137Cs, divided by the ratio of activities of the nuclides in the source term. The 90Sr/137Cs ratios showed a slight decreasing tendency with depth [11], though the number of data available was limited. As Koma et al. [12] proposed, the transport ratio may not provide information on the transport process, but may merely describe the difference between the activities of the initial (fuel debris as a source term) and resulting (contaminated) materials. Thus, it is essential to know, or presume, contamination behavior in a variety of off-site and on-site radioactive wastes. Koma et al. analyzed the transport ratio based on the reported values of 137Cs and 90Sr concentrations in the contaminated soil sampled at several points across the FDNPS [13] and surrounding the FDNPS [14, 15]. The transport ratio might depend on the direction from the reactor buildings because the source term of the reactor fuel, the release conditions of the nuclides, and the timing of the hydrogen explosion were different. Nevertheless, the ratios seem to be distributed in one or two orders of variation, probably due to the complex diffusion of nuclides released into the environment. For a more reliable estimate of the total amounts of wet and dry depositions, analysis of the transport ratio is still under discussion. Moreover, the impact of resuspension corresponding to a soil-dust suspension (as an atmospheric carrier) on the temporal change of the radionuclide concentration in the surface soil has long been discussed [16]. In the area near FDNPS after the accident, Steinhauser et al. [17] found that a significant intermittent release of airborne radionuclides in 2013, long after the initial huge releases in spring 2011. Since this process would also change the transport ratio over a long period of time, the high inventory of 137Cs and 90Sr as a source term should be analyzed early after the accident.
Even more than 5 years after the accident, radionuclide transport into deeper soil layers would be expected to proceed in each redistribution process. In particular, the depth profile of contamination is one of the most important data required to develop a model of nuclide migration. In 2011, Tanaka et al. [18] investigated vertical profiles of 137Cs (and 131I) in the core soil samples collected about 60 km from the FDNPS, and found that more than 87% of Cs distributed at depths of 0–5 cm . Takahashi et al. [19] investigated the vertical distribution of 137Cs in soil profiles to characterize the patterns of migration in different types of land use, such as forest, grassland, and agriculture. Such land use patterns have a large influence on some soil properties and on the migration processes of 137Cs above ground, resulting in different vertical distributions of 137Cs. This might be explained by a trend of exponential decline with depth, but the vertical distribution in the soil column of radionuclides other than Cs has not yet been studied systematically. With the object of on-site dose reduction to improve the work environment of the site, a decontamination treatment, including tree trimming, removal of surface soil, deep plowing, and asphalt placement, has already been completed. Therefore, it was difficult to collect highly contaminated soil at the site, and the objective of this study was to measure the high radioactivity concentrations of Sr and Cs in soil samples collected in the vicinity of the FDNPS. This knowledge would provide an insight into understanding the depth profile of radionuclides in soil.
Additionally, clarification of the role of organic matter in the distribution of 137Cs and 90Sr in soil would be helpful to understand the chemical forms of nuclides and the interactions between soil–water phases. Sequential extraction methods have been performed to determine radionuclides in soil samples [20,21,22], but a domestic or international standard procedure has not been defined yet. Therefore, a multi-step extraction scheme for the present study was modified by referring to previous studies [23], in which the extractants used at each stage are generally intended to release the nuclides associated with particular soil phases, such as water-soluble, ion-exchangeable, acid-soluble, reducible, oxidizable, and residual phases.
In addition, the sorption distribution coefficient, Kd, of nuclides on the soils, collected at different depths from the ground surface, was determined by laboratory batch sorption experiments, for use as one of the fixed parameters in a model to predict future long-term vertical transport.
Experimental
Sampling protocol
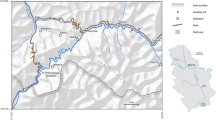
As shown in Fig. 1, two undisturbed sampling sites were selected at the site border of FDNPS, at 37º 24′ 49.84″ N and 141º 01′ 44.18″ E (called ‘site-A’) and at 37º 25′ 01.38″ N and 141º 01′ 22.67″ E (called ‘site-B’). Three samples were collected at each site, in January 2016, August 2016, and June 2017, and are denoted as site-A-1601, site-A-1608, site-A-1706, site-B-1601, site-B-1608, and site-B-1706. The current vegetation in site-A and that in site-B are different from each other. The site-A is a sparse coniferous forest of Cryptomeria japonica, Pinus thunbergii, Pinus densiflora, and Abies firma, covered with bamboo grass and other undergrowth. The site-B is a mixed forest of deciduous broad-leaved (Cerasus spp. and Alnus firma) and evergreen coniferous trees (Cryptomeria japonica and Pinus thunbergii) in approximately equal numbers. The tree height and tree density of the canopy layer in site-B are 12–18 m and 1,460 ha−1, respectively. Evergreen broad-leaved shrub trees (Eurya japonica and Neolitsea sericea) exist sparsely as undergrowth cover.

Map of soil sampling sites (sites-A and -B) in the vicinity of FDNPS
These areas were contaminated at a level of 82 µSv h−1 for site-A and 138 µSv h−1 for site-B (as of January 2016), at a height of 5 cm from the ground surface. Fresh brownish soil at sites-A and -B was classified as Andosol. The thickness of the organic litter layer at each site was a few centimeters on average. After removing this layer by hand, the soil was sub-sectioned into 1 cm increments for the uppermost 20 cm by using a scraper plate (450 cm2 in surface area) with careful attention to cross-contamination [24].
For the sorption experiment, natural spring water was collected as the aqueous phase on the slope of the small forest near the site-A sampling point. The water was kept in the refrigerator until use. One liter of water was equilibrated with 20 g fresh soil at 10 cm-depth at site-B, followed by filtration using a PTFE membrane with a 0.45 μm pore size (Advantec) attached to a plastic syringe. This solution is referred to as “soil water” in this study. The chemical properties of the fresh spring water and the soil water were measured by ICP-OES (SpectroBlue, Spectro Analytical Instruments) for major elements, and with an electrical conductivity meter (CM-31P, DKK-TOA).
Sample preparation and characterization
The soil was passed through a 10-mesh sieve (passing particles smaller than 2 mm) to exclude large fallen leaves, branches, and pebbles. After careful homogenization by hand, the soil sample at each depth was dried in air at 105 °C overnight, its density then being defined as the dry bulk density (g cm−3). The moisture content, which is a measure of the amount of water contained in unit mass of soil, was estimated. Standard grinding using a tungsten carbide mortar, and mounting the soil on a 0.5 mm thick glass slide, was performed prior to X-ray powder diffraction (XRD) analyses. The XRD measurements were performed with an Ultima IV X-ray diffractometer (RIGAKU SmartLab) using Cu K-α1 radiation at 45 kV and 200 mA with a D/teX Ultra 250 detection system. The range of 2θ scanning was from 4° to 90° in steps of 0.01° with a counting time of 0.06 s per step.
For gamma-ray measurement of 137Cs, the soil was placed in a 100-mL U-8 polystyrene cylindrical container 50 mm in inner diameter (a standard container in Japan), and the weight and height were measured in the container. The fresh dry soils sampled at four depths (1, 4, 9, and 17 cm) were used for the sequential extraction experiment.
For ICP-MS measurement of 90Sr, we considered that the simple digestion method should be suitable for leaching Fukushima-derived 90Sr sorbed on the surface of soil mineral particles and complexed with natural organic matter, although this method would not allow the dissolution of all mineral components of the soil, and might result in the partial extraction of natural strontium. The dried samples were burnt in a porcelain crucible at 500 °C for 4 h in a muffle furnace (FO100, Yamato), and natural organic matter was estimated as the weight loss as loss on ignition. Following ignition, a 4.5 g portion of the soil placed in a closed Teflon vessel was digested with 15 ml of 20% (v/v) nitric acid at 200 °C for 2 h under a weakly pressurized condition. The resulting suspension was centrifuged at 2,000 × g for 5 min, and the supernatant solution was passed through a 0.45 μm membrane filter (ADVANTEC).
Determination of Cs and Sr radioactivity concentrations
The 137Cs radioactivity concentration was measured using a gamma-ray spectrometry system consisting of an n-type coaxial high-purity Ge detector with a Be window (GMX15P4, Ortec, relative efficiency 15%) coupled to a PC-based 8 K multichannel analyzer (Trump ISA-8 K, Ortec). The energy resolution measured in terms of full width at half maximum (FWHM) is 1.84 keV at 1332.5 keV of 60Co gamma ray energy. The detection efficiency of the gamma rays was calibrated with an accuracy of 2–10% using a multiple gamma ray standard source (MX033U8PP, Japan Radioisotope Association) with an energy range of 88 keV to 1836 keV. The spectrum was acquired for 137Cs (662 keV), and the radioactivity was normalized by the dried soil weight.
The concentration of 90Sr in soil was determined by the conventional ICP-MS method as described by Takagai et al. [25]. Briefly, a mass spectrometer (ELAN DRC II, PerkinElmer) was equipped with the following accessories: (1) an on-line chelate column (50 mm long and 4 mm in inner-diameter) filled with extraction chromatographic resin (0.2 g of Sr resin, Eichrom) to separate strontium from most other metal ions, connected to a peristaltic pump for 8-channel flow (FIAS 400, PerkinElmer); (2) an ultrasonic nebulizer (U5000AT, CETAC) to enhance the sensitivity; and (3) a collision-reaction cell (the so-called dynamic reaction cell system preinstalled in ELAN DRC II) to separate the isobars 90Zr and 90Y from 90Sr by selective reaction of Zr and Y with ultrapure O2 gas (> 99.9999%, 1.6 mL min−1). The Sr resin, which contains hydrophobic 18-crown-6 (crown ether) derivative in 1-octanol on an inert polymeric support, can selectively retain strontium, and the matrix ions such as Na+ and most of the minor elements can be eluted from the resin by 20% (v/v) nitric acid. The concentrated strontium was leached out using 10 mL of 0.01 M nitric acid, and the ICP-MS measurement was then carried out. The extremely low molar concentration of 90Sr was determined based on the peak area of counting with a calibration curve of non-radioactive Sr standard solutions, and with yield corrections applied by using the 115In internal standard, and 86Sr, which is the one of isotopes leached from the sample soils, where the matrix 86Sr was diluted below the upper detection limit of ICP-MS. In this solution condition, a tailing interference from a neighboring peak of 88SrH was also negligible. Thus, the instrumental settings and on-line chemical conditions were optimized. Radioactivity measurement series No. 2 [26] was applied to some of the soil samples to determine the specific radioactivity concentration of 90Sr. Shortly, the soil sample was ashed, and then nitric acid was added to soil ash in the digestion process. 90Sr was isolated from 90Y by co-precipitation methods using oxalic acid, Ba(II), and Fe(III). The radioactivity of 90Sr in soil was determined by a gas flow detector after milking of 90Y. The data obtained by both ICP-MS and radioactivity measurement series No.2 were used to discuss the distribution of 90Sr in the depth profile.
Sequential extraction procedure
Three steps of sequential treatment were each divided into four targets: (1) ultra-pure water at neutral pH to extract water-soluble Cs and Sr under typical mild conditions; (2) ammonium acetate at neutral pH to exchange weakly bound cations; (3) hydrogen peroxide in hot acid medium to extract these cations bound to the soil surface and the organic component; and subsequently; 4) Cs and Sr in the remaining residue. Fig. S1 defines the four fractions in the partitioning of 137Cs and 90Sr, and some details of the solution conditions, contact times, and treatment methods.
A 25 g-dry weight portion of fresh soil (site-A-1706 or site-B-1706, sampled at 1, 4, 9, or 17 cm in depth) was treated with 62.5 mL pure water in a 250 mL perfluoro alkoxyalkane (PFA) bottle. During the extraction under atmospheric conditions at 25 °C, the bottles were shaken vigorously at 70 oscillations per minute using a shaker (BR-43FL-MR, Taitec) for 1 h. The supernatant obtained by a brief centrifugation treatment followed by passing through a 0.45 μm polytetrafluoroethylene (PTFE) membrane filter was treated as fraction–W. The residue was washed with an additional 10 mL of pure water, and the filtrate was combined with fraction–W.
A 100 mL portion of 1 M ammonium acetate at pH 7 was added to the residual soil. After 16 h of shaking, the supernatant contained cations exchangeable by ammonium ions. It was filtered through a 0.45 μm pore membrane filter, and the filtrate was treated as fraction–E.
The residue was added to 50 mL of H2O2 solution (9:1v/v 30% H2O2: 0.1 M HNO3) in a 250 mL PFA bottle. The suspension was kept without shaking at room temperature to avoid the uncontrolled acceleration of an exothermic decomposition reaction with the large amount of organic matter in the soil. The solution was then heated for 4 h at 85 °C in a hot water bath, and the supernatant was separated from the soil. The extraction procedure with H2O2 + HNO3 was repeated once more with 16 h aging at 85 °C. After separation of the supernatant from the soil, 100 mL of 1 M ammonium acetate (pH 5 with HNO3/CH3COOH) was added to the supernatant and incubated for 16 h at room temperature. The supernatants were combined and filtered through a 0.45 μm pore membrane filter, and were denoted as fraction–O, which provides a measure of 90Sr and 137Cs bound to organic matter. The residual soil minerals that were tightly bound to 90Sr and 137Cs were dried as fraction–R.
Fractions-W, -E, and -O in polypropylene tubes were evaporated using a dry block bath (EB-303A, AZ-ONE), and the radioactivity of 137Cs was determined using gamma-ray spectrometry. The residue from evaporation in the tube was dissolved again by 10 mL of 20 wt% nitric acid, followed by extraction of strontium overnight with 1 g of the Sr resin. After stripping the Sr from the resin with pure water, 90Sr was measured by ICP-MS as described above. The 90Sr radioactivity of fraction–R was estimated by subtracting the sum of the 90Sr activities of fractions-W, -E, and -O from the total radioactivity.
Batch experimental study of the sorption coefficients
The experimental studies on adsorption of Cs and Sr were carried out in a batch system. A 0.2 g portion of each of the specimens (site-A-1706 and site-B-1706) without decomposition of the soil organic matter was mixed with 10 mL of the soil water as aqueous phase in a 13-mL polypropylene (PP) centrifuge tube (Sarstedt), and the initial 85Sr and 134Cs radioactive concentrations were each set to ca. 20 kBq by the addition of carrier-free solutions. This radioactivity corresponds to ca. 3 × 10−11 and 3 × 10−10 mol dm−3 (M) Sr and Cs, respectively. Since the maximum inventory of 134Cs in the highly contaminated surface soils was ca. 0.1–0.5 kBq per 0.2 g, the initial radioactivity lower than the added 20 kBq was negligible impact on the present sorption study.
The initial pH was adjusted by addition of HCl or NaOH solutions. The pH (D-52/9611-10D, Horiba) and electroconductivity (WM-32EP/CT-27112B, TOA-DKK) were recorded. During the batch experiments under atmospheric conditions at 25 ± 1 °C, the centrifuge tubes were agitated using a bench-top rolling shaker at a speed of 6 rpm (Roller 10, IKA) for 7 d. The supernatant was filtered through a 0.45-μm pore filter membrane, followed by transfer to a new 13-mL PP test tube. The filtrate was acidified by addition of a tiny portion of conc. HCl, and evaporated using an aluminum block heater (EB-303A, AS One) at 368 K to prepare a solidified point-like source of gamma rays. All the chemical reagents used were of analytical grade and were used without further purification.
The photo peak area corresponding to each nuclide (605 keV for 134Cs and 514 keV for 85Sr) was evaluated, making use of calibration curves prepared by using point-like sources of original tracer solutions. The sorption distribution coefficient Kd (m3 kg−1) of radionuclides was calculated using Eq. (1),
where V is the volume (m3) of aqueous solution at sorption equilibration, W is the weight (kg) of the air-dried soil sample, and A0 and A are, respectively, the initial and final (after equilibration) activities in the aqueous phase. The desorption behavior of the sorbed 137Cs was examined using a sample of soil from site-B at 1 cm depth without adding the radioactive tracer of Cs. The Kd values were evaluated based on Eq. (1) and the initial radioactivity in the soil.
Results and discussion
Soil properties
Figure 2 shows the depth profiles of the dry bulk density and the moisture content. The bulk density increased significantly with soil depth between 0 and 5 cm, since the lower layers are generally compacted and have less organic matter, less aggregation, and less root penetration compared to the surface layers. On the other hand, the water content of the topsoil at 0-5 cm depth was higher than 0.5, and decreased with depth. A similar trend for the dry bulk density and the water content with depth is shown in the tables of references [27, 28]. A decrease in the loss on ignition with depth is also a feature of the soils, as shown in Fig. 2. However, below the depth of 5 cm (except site-B-1601), the loss on ignition was practically of the same low order at the different depths, indicating very little organic matter. This tendency has been found in the previous work, which was done in a forest site of the southwestern part of Fukushima city [29]. Thus, these three major properties of soil in Fig. 2 are closely linked.

Depth profile of dry bulk density, moisture content, and decrease in the loss on ignition of soil samples
The XRD spectra of soils site-A-1706 and site-B-1706 at four depths are shown in Fig. S2. The predominant mineral fractions of soil were found to be SiO2 (quartz, cristobalite), plagioclase (albite), which might be a product of decomposed granite, and vermiculite. The clay component was not clearly assigned, but it could be generally composed of fertile soil. The XRD pattern was independent of the depth up to 20 cm, indicating a similar mineral composition.
Vertical distribution of 137Cs
The vertical distributions of the 137Cs concentration at site-A and site-B in the vicinity of the FDNPS are shown in Fig. 3. The activity concentrations were corrected to allow for the different sampling dates. Some experimental data (site-A-1608 in Fig. 3) could not be obtained because large stones and roots at the depth prevented further digging. The row value data are listed in Table S1 as supplemental information. According to a general gamma-ray spectroscopic measurement [30], the 137Cs activity of the surface soil samples at 0–5 cm depth was found to be as high as 1.07 × 107 Bq kg−1 dry weight for site-B. The concentration of 137Cs decreased with depth, but the concentration below 10 cm in depth was still two orders of magnitude lower than that in the top-soil even 5 years after the initial fallout. This suggests that some chemical components, such as clay minerals and natural organic matter, interact strongly with radioactive cesium [31]. The sorption would be highly selective for Cs, and this is supported by microscopic studies of cesium sorption on minerals such as clay [32], and biotite and muscovite [33]. A similar tendency for exponential distributions has been found in the previous work of the MEXT project, which was done in a wide area of 100 km radius around the FDNPS in 2011–2012 [8]. The maximum 137Cs concentration at the soil surface was ca. 104 Bq kg−1, which is three orders lower than our findings in 2016 at different distances from the FDNPS. Those researchers also found an anomalous temporal change in distribution in some sampling regions due to soil disturbance, cultivation, or decontamination work [34]. Kurikami et al. [35] tested a modified diffusion-sorption-fixation model for reproducing radiocesium depth profiles measured in Fukushima soils. The present study also found that the long-tail exponential distribution was well maintained in undisturbed soils. Nevertheless, three depth profiles in sampling points site-A and site-B do not exhibit exactly the same temporal change to deeper migration. Even if soil sampling was done in an area with a radius of a few meters, the detailed conditions of soils, which might affect the migration, would be different from each other. This fact would also apply to other radionuclides.

Vertical distribution in soils of 137Cs (upper) and 90Sr (bottom). The symbol * denotes the results measured by the method in radioactivity measurement series No. 2 [26]. The gray plots represent the values below LOD for ICP-MS
Vertical distribution of 90Sr
The radioactivity concentrations of 90Sr measured by ICP-MS are plotted, with some results from radioactivity measurement series No. 2, in Fig. 3. The values for recovery yield correction in the mass spectrometry including the on-line extraction chromatography was 30% in average but scattered depending on samples. Therefore, the 90Sr concentration was corrected by using each recovery yield. Although there are not enough data to verify the integrity of the values evaluated by both analytical methods, there appears to be reasonable consistency, within the variability of the digits, in the upper soil layers. Therefore, both data can be treated and considered in this study. The row value data are also listed in Table S1. The activity concentration of 90Sr ranges from hundreds to 10,000 Bq kg−1, which is distinctly high in comparison to the background level of less than ca. 10 Bq kg−1 (in average) due to the legacy of global fallout before the severe accident at the FDNPS [1]. The values for 90Sr are lower than those for 137Cs, and this fact would be due to the lower volatility of Sr from the source terms known as melted cores.
90Sr data at 500 m in the southwestern direction from the Units 1 and 2 exhaust stacks, which are located close to site-A, were reported by TEPCO in 2011 [36]. These values are relatively low, in the range between (4.0 ± 0.17) × 101 and (5.7 ± 0.06) × 102 Bq kg−1_dry soil, compared to the maximum values of 1.7 × 103 and 1.4 × 104 Bq kg−1 fresh soil at site-A and site-B, respectively, as seen in Fig. 3. This indicates that directional plumes deposit radionuclides around these areas.
Unlike that of 137Cs retained in the surface layer, the vertical distribution of the 90Sr concentration is relatively “flat”, implying that strontium could diffuse into the soil, although some values in the deeper and even shallower layers were below the limit of detection (LOD, 3 sigma) for ICP-MS. Here, the LOD value was not the same for each sample, because the calibration curve was obtained in every measurement. In particular, the strong affinity of clay minerals in the top soil for Cs has been analyzed by various experimental and theoretical studies [37, 38]. The 90Sr depth profile of site-B samples showed a slightly decreasing tendency with depth, and a maximum concentration at the surface, similar to the Cs profile. On the other hand, the profile of the site-A samples has a small peak for layers at a depth of ca. 5 cm or deeper. The continuity in depth of the bulk properties, such as the loss on ignition, was found as discussed in the previous section, supporting the absence of any clear difference in the properties of all soil samples. Therefore, other possible reasons, such as the formation of specific colloids, the presence of organic matter and particles, and the influence of the aquifer on horizontal transport, need to be considered in further studies.
Sequential extraction
Four soil samples at depths of 1, 4, 9, and 17 cm were examined. The pH values of fraction-W after contacting with soil for 1 h were 5.2 (1 cm), 4.6 (4 cm), 4.6 (9 cm), and 4.6 (17 cm) for site-A, and 5.7 (1 cm), 4.0 (4 cm), 4.4 (9 cm), and 4.6 (17 cm) for site-B, respectively. These soil pH data, except those in the top soil, were less than 5, which could be categorized as strongly acidic soil.
The activity concentrations of 137Cs released in sequential extraction treatments at the four depths are shown in Fig. 4. Fraction-R for 137Cs in the surface soils was predominant (> 99%) in the present sequential extraction system, indicating a strong sorption of most Cs on the soil, whereas the 137Cs in fractions-W, -E, and -O are quite minor. It is noted that the fraction-W of Cs was only found in the top soil of site-A and site-B containing a richer litter. On the contrary, the existence ratio of 137Cs in fractions-E and -O increased with depth. The Cs component of these fractions was mobile in the soil compared to that of fraction-R. It can be concluded that the proportion of mobile components relative to fraction-R increased with depth, but most of the Cs in the deeper soil layers still existed in fraction-R.

137Cs and 90Sr fractionations of soil samples at site-A-1706 and site-B-1706 obtained by sequential extraction
The results of sequential extraction for 90Sr are shown in Fig. 4. Though some 90Sr concentrations in deeper layers were low, close to or below the LOD for ICP-MS, the major fraction of 90Sr would be fraction-R, suggesting a strong interaction with soil at site-A and site-B, similar to the 137Cs system. However, the proportion of mobile components, as in the individual fractions-W, -E, and -O, relative to fraction-R, is higher than that shown by Cs. Such a difference in the sequential extraction could result in the vertical distribution of 137Cs and 90Sr. But in particular, fraction-E in the top soil of site-B (containing one of the highest 90Sr concentrations in this study) is as high as fraction-R, though the sorption and desorption mechanism is still not completely understood.
Adsorption properties of 134Cs and 85Sr tracers
The major elements in the fresh spring water and the soil water were measured using ICP-OES. The respective concentrations (spring/soil) are 15.2/13.8 ppm for Na, 0.8/4.4 ppm for K, 2.6/0.8 ppm for Mg, 1.7/0.8 ppm for Ca, 11.4/13.2 ppm for Si, and ND/0.2 ppm for Al. The electrical conductivities were 13 and 34 mS m−1, respectively, which is typical of the natural groundwater system in Japan. Thus, there is a slight increase in the number of major ions, but the change is not significant.
Figure 5 shows the pH dependence of log Kd (m3 kg−1) for tracer 134Cs between the surface soil of site-B and the soil water. The Kd value decreased with a gradual decrease in pH from 7 to 3.5. A similar trend with pH is shown in the plots of references [39,40,41,42]. These results may suggest a tendency for surface complexation at a mineral sorption site or with organic matter. The desorption experiment was performed using the top-soil (1 cm) of site-B, as shown in Fig. 5. The log Kd values were as high as 2 in the pH range of 3–6, suggesting that the existing sorbed 137Cs could not be released easily from soil, especially at specific binding sites of clay mineral particles. With regard to the contrast between the sorption and desorption values, the additional tracer 134Cs may be sorbed not only on the specific and strong sorption sites but also on weaker sorption sites. Such a strong interaction of Cs with clay minerals in soil has long been studied [43]. The Kd values for site-B at depths of 4, 9, and 17 cm, and those for site-A are also plotted. The log Kd values for Cs on site-B soil increased from 0 (1 cm) to 1 (17 cm). The values and the depth dependence for site-A are almost the same as for site-B, supporting the similarity of soil properties such as the content of organic matter and minerals and their variation with depth.

pH and depth dependences of Kd values for 134Cs and 85Sr tracers, with reference values
The pH dependence of Kd for 85Sr is also shown in Fig. 5. It is noted that log Kd for site-B increased with increasing pH, similar to the Cs system, with values slightly higher than those for Cs. The different mineral components and contents of other compounds, such as organic matter, might also be sensitive to the range of Kd values. Meanwhile, the log Kd values are independent of the depth. The clear difference between Cs and Sr could be caused by the different sorption properties of different types of sites in soil.
A specific interaction of the Sr2+ ion with a certain negatively charged site on some mineral component of the present soils is considered. The Sr sorption onto the minerals has been investigated. In the early study by Zachara et al. [44], the sorption on calcite as a surface exchange reaction between Sr2+ and Ca2+ (CaCO3) was negligible. But, it would not be applied to the present study, since no significant content of calcite was found in the soil samples collected near FNDPS. Bracco et al. [45] suggested that Sr was not only sorbed but also incorporated to barite surface (BaSO4), probably due to the similar ion size and chemical properties of alkaline earth metals. Therefore, the effect of these reactions on the 90Sr sorption would be negligible.
The pH dependence for Sr has been generally recognized for clay converted to the sodium form [46], and zeolite [47]. Moreover, a sorption reaction was observed in amorphous silica and in the goethite system [48]. In a higher neutral pH region, Konishi et al. [42] measured the Kd values at neutral pH for the loam (a mixture of clay and silt) collected in Tohoku district (including the Fukushima region), Japan. The log Kd plots seem to be relatively low compared with our results, which might be due to a higher initial Sr concentration (10−5 M). However, the log Kd values for tracer amounts of radioactive 90Sr in silty-clay loam in Savar, Bangladesh at pH 8 [41] were in good agreement with the data of Konishi [42]. For a future simplified modeling of the interaction between 90Sr and the Fukushima soil surface, the apparent surface charge of the soil may be primarily assumed to result from a simple acid–base reaction at a mineral surface: ≡ SOH2+ ↔≡ SOH + H+ and ≡ SOH ↔ ≡ SO– + H+, where ≡ SO− denotes a sorption reaction site on a mineral or a functional group of an organic compound, and the number of sites increases with increasing pH. Trace amounts of 85Sr sorption would then result from the following reaction: ≡ SOH + Sr2+ ↔ ≡ SOSr+ + H+. Further studies should be needed to clarify the macroscopic interaction of 90Sr.
Transport ratio of 90Sr
The transport ratio TR,x, of 90Sr is defined as
where I [Bq] is the inventory of 137Cs and 90Sr in the spent fuel of the unit from which the deposited radionuclides were released, and CCs,x and CSr,x are the activity concentrations (Bq kg−1) of the x cm thick soil sample (from the surface). Koma et al. [12] reported the TR of the surface soil collected in the southern area of the plant. Some values in the range of 10−3 to 10−4 gradually decreased with time, which might suggest that Sr migrated to the deeper soil layers ahead of Cs. Figure 6 shows the relationship of CCs,1 and CSr,1 (x = 1) with reference values, which were analyzed for the surface soil samples. Sahoo’s data obtained in the south direction [49] are similar to the present values, whereas those in the north-west direction are lower, and the C values of 90Sr are relatively higher, suggesting a higher TR value. The ratio of CCs to CSr seems to have a good linear relationship with the results of this study.

Relationship of CCs,1 and CSr,1 with reference values
Based on the depth profile in the present study, the calculated TR,x values (x = 1, 2, …, 20) are shown in Fig. 7, where ISr,fuel/ICs,fuel is assumed to be 1.335 [50]. It was found that most of the TR,x gradually increased with depth because of the increase in the ratio of CSr to CCs in the deeper layers. Similar to the existing data [12], most of the TR,x values fell between ca. 10−3 and 10−4 regardless of the soil layer thickness. However, the TR,x of site-A-1608 increased sharply in the layers between the surface and 6 cm depth, which was caused by a significant increase in the 90Sr concentration. The interpretation of such a local increase in concentration in specific layers may require consideration of additional transport processes such as suspended matter and horizontal migration.

Depth dependence of transport ratio TR,x of 90Sr
Conclusions
The depth profile of 90Sr in soil samples collected from the surface to a depth of 20 cm near the FDNPS was investigated by ICP-MS with a chemical separation system dedicated to Sr concentration. The inventory of 90Sr deposition on the soil was much less than that of 137Cs because of its lower volatility in the melted cores from the severe accident. In the 5 years after deposition, 90Sr migrated to deeper soil layers, and some local peaks were observed, suggesting a significant vertical migration. On the other hand, the strong sorption of 137Cs on the surface soil component was found to be the same as previously reported. The batch-wise sorption experiment at the neutral pH of the natural water system using carrier-free isotopes of 85Sr and 134Cs showed that the logarithmic apparent adsorption coefficients of these nuclides fell within the same range of 0–1. In addition, the sequential extraction showed that the predominant sorption species of 90Sr were not only soil minerals but also organic compounds, whereas 137Cs could be strongly sorbed on specific minerals such as clay. As a consequence, 137Cs in forest soils has migrated very slowly, and the dramatic downward migration of large amounts of 137Cs would be unlikely to be found in the future. Such depth profiling suggests that high concentrations of Cs and Sr were initially deposited on the surface litter and soil, and were then gradually eluted by ion exchange reactions to be transferred to deeper soil layers. With the knowledge of the present distribution, further investigation of model parameters, such as the effective diffusion coefficient, constant flow velocity, and the effective first-order rate coefficient for ion exchange between the aqueous domains, is needed for reliable modeling in the future.
References
Results of the radiation monitoring of soil in Fukushima Prefecture (2012) Disaster provision main office of Fukushima Prefecture. https://radioactivity.nsr.go.jp/en/contents/6000/5025/24/232_e_0409.pdf. Accessed 17 June 2020
Povinec P, Hirose K, Aoyama M (2013) Fukushima accident: radioactivity impact on the environment. Elsevier, Amsterdam. ISBN 978-0-12-408132-1
Zheng J, Tagami K, Bu W, Uchida S, Watanabe Y, Kubota Y, Fuma S, Ihara S (2014) 135Cs/137Cs isotopic ratio as a new tracer of radiocesium released from the Fukushima nuclear accident. Environ Sci Technol 48:5433–5438. https://doi.org/10.1021/es500403h
Takahashi T (ed) (2016) Radiological issues for Fukushima’s revitalized future. Springer, Tokyo, p 232. https://doi.org/10.1007/978-4-431-55848-4
Nakanishi TM, Kobayashi NI, Tanoi K (2013) Radioactive cesium deposition on rice, wheat, peach tree and soil after nuclear accident in Fukushima. J Radioanal Nucl Chem 296:985–989. https://doi.org/10.1007/s10967-012-2154-7
Hirose K (2012) Fukushima Dai-ichi nuclear power plant accident: summary of regional radioactive deposition monitoring results. J Environ Radioact 111:13–17. https://doi.org/10.1016/j.jenvrad.2011.09.003
Sanada Y, Torii T (2015) Aerial radiation monitoring around the Fukushima Dai-ichi nuclear power plant using and unmanned helicopter. J Environ Radioact 139:294–299. https://doi.org/10.1016/j.jenvrad.2014.06.027
Results of the nuclide analysis of soil sampling at 2,200 locations in Fukushima Prefecture and neighboring Prefectures (decay correction: June 14, 2011) (Sr-89, Sr-90) (2011) Ministry of Education, Culture, Sports, Science and Technology (MEXT), and the Secretariat of the Nuclear Regulation Authority-Japan. https://emdb.jaea.go.jp/emdb/en/portals/1020101001/. Accessed 17 June 2020
Radiation monitoring of soils in Fukushima Prefecture (in Japanese), https://www.pref.fukushima.lg.jp/sec_file/monitoring/etc/dojou120406.pdf. Accessed 17 June 2020
Steinhauser G, Schauer V, Shozugawa K (2013) Concentration of strontium-90 at selected hot spots in Japan. PLoS ONE 8:e57760. https://doi.org/10.1371/journal.pone.0057760
Rosenberg BL, Ball JE, Shozugawa K, Korschinek G, Hori M, Nanba K, Johnson TE, Brandl A, Steinhauser G (2017) Radionuclide pollution inside the Fukushima Daiichi exclusion zone, part 1: depth profiles of radiocesium and strontium-90 in soil. Appl Geochem 85:201–208. https://doi.org/10.1016/j.apgeochem.2017.06.003
Koma Y, Shibata A, Ashida T (2017) Radioactive contamination of several materials following the Fukushima Daiichi nuclear power station accident. Nucl Mater Energy 10:35–41. https://doi.org/10.1016/j.nme.2016.08.015
Tokyo Electric Power Company, Incorporated (TEPCO) (2011) Detection of radioactive material in the soil in Fukushima Daiichi Nuclear Power Station. http://www.tepco.co.jp/en/press/corp-com/release/11032812-e.html. Accessed 17 June 2020
Committee on the construction of maps for radiation dose distribution, concerning analytical result of soil (Cesium 134 and 137), 2011 Document No. 7–1, 29 August 2011 (in Japanese). http://www.mext.go.jp/b_menu/shingi/chousa/gijyutu/017/shiryo/1310688.htm. Accessed 17 June 2020
Committee on the construction of maps for radiation dose distribution, concerning analytical result of radionuclides in soil (Strontium89, strontium90, plutonium238, plutonium239 + 240) (2011) Document No. 10-1 (for reference), 30 Sep 2011 (in Japanese). http://www.mext.go.jp/b_menu/shingi/chousa/gijyutu/017/shiryo/1311753.htm. Accessed 17 June 2020
Igarashi Y, Aoyama M, Hirose K, Miyao T, Nemoto K, Tomita M, Fujikawa T (2003) Resuspension: decadal monitoring time series of the anthropogenic radioactivity deposition in Japan. J Radiat Res 44:319–328. https://doi.org/10.1269/jrr.44.319
Steinhauser G, Niisoe T, Harada KH, Shozugawa K, Schneider S, Synal H-A, Walther C, Christl M, Nanba K, Ishikawa H, Koizumi A (2015) Post-accident sporadic releases of airborne radionuclides from the Fukushima Daiichi nuclear power plant site. Environ Sci Technol 49:14028–14035. https://doi.org/10.1021/acs.est.5b03155
Tanaka K, Takahashi Y, Sakaguchi A, Umeo M, Hayakawa S, Tanida H, Saito T, Kanai Y (2012) Vertical profiles of iodine-131 and cesium-137 in soils in Fukushima prefecture related to the Fukushima daiichi nuclear power station accident. Geochem J 46:73–76. https://doi.org/10.2343/geochemj.1.0137
Takahashi J, Tamura K, Suda T, Matsumura R, Onda Y (2015) Vertical distribution and temporal changes of 137Cs in soil profiles under various land uses after the Fukushima Daiichi nuclear power plant accident. J Environ Radioact 139:351–361. https://doi.org/10.1016/j.jenvrad.2014.07.004
Shand CA, Cheshire MV, Smith S, Vidal M, Rauret G (1994) Distribution of radiacaesium in organic soils. J Environ Radioact 23:285–302. https://doi.org/10.1016/0265-931X(94)90067-1
Rigol A, Roig M, Vidal M, Rauret G (1999) Sequential extractions for the study of radiocesium and radiostrontium dynamics in mineral and organic soils from Western Europe and Chernobyl areas. Environ Sci Technol 33:887–895. https://doi.org/10.1021/es980720u
Rauret G, Firsakova S, European Commission, Brussels (eds) (1996) The transfer of radionuclides through the terrestrial environment to agricultural products, including the evaluation of agrochemical practices, Experimental collaboration project No. 2 (1996). ISBN 92-827-5193-7
Ure AM, Davidson CM (eds) (2002) Chemical speciation in the environment 2nd edn. Blackwell Science, Glasgow, 265-300 (Chapter 10). ISBN 0-632-05848-X
Loughran RJ, Wallbrink PJ, Walling DE, Appleby PG (2003) Sampling methods. In: Zapata, F. (ed) Handbook for the assessment of soil erosion and sedimentation using environmental radionuclides, pp 41–57 (Chapter 3). ISBN 978-1-4020-1041-5
Takagai Y, Furukawa M, Kameo Y, Suzuki K (2014) Sequential inductively coupled plasma quadrupole mass-spectrometric quantification of radioactive strontium-90 incorporating cascade separation steps for radioactive contamination rapid survey. Anal Methods 6:355–362. https://doi.org/10.1039/C3AY41067F
MEXT, Radioactivity measurement series No. 2: Environmental sample collection methods (2003) Japan Chemical Analysis Center, Japan [in Japanese]. https://www.kankyo-hoshano.go.jp/series/lib/No2.pdf
Takahashi J, Onda Y, Hihara D, Tamura K (2019) Six-year monitoring of the vertical distribution of radiocesium in three forest soils after the Fukushima Daiichi nuclear power plant accident. J Environ Radioact 192:172–180. https://doi.org/10.1016/j.jenvrad.2018.06.015
Muto K, Atarashi-Andoh M, Matsunaga T, Koarashi J (2019) Characterizing vertical migration of 137Cs in organic layer and mineral soil in Japanese forests: four-year observation and model analysis. J Environ Radioact 208–209:106040. https://doi.org/10.1016/j.jenvrad.2019.106040
Koarashi J, Nishimura S, Atarashi-Andoh M, Muto K, Matsunaga T (2019) A new perspective on the 137Cs retention mechanism in surface soils during the early stage after the Fukushima nuclear accident. Sci Rep 9:7034. https://doi.org/10.1038/s41598-019-43499-7
Dohi T, Ohmura Y, Kashiwadani H, Fujiwara K, Sakamoto Y, Iijima K (2015) Radiocaesium activity concentrations in parmelioid lichens within a 60 km radius of the Fukushima Daiichi nuclear power plant. J Environ Radioact 146:125–133. https://doi.org/10.1016/j.jenvrad.2015.04.013
Cremers A, Elsen A, De Preter P, Maes A (1988) Quantitative analysis of radiocaesium retention in soils. Nature 335:247–249
Liu X, Lu X, Wang R, Zhou H (2004) Effects of layer-charge distribution on the thermodynamic and microscopic properties of Cs-smectite. Geochim Cosmochim Acta 72:1837–1847. https://doi.org/10.1016/j.gca.2008.01.028
McKinley JP, Zachara JM, Heald SM, Dohnalkova A, Newville MG, Sutton SR (2004) Microscale distribution of cesium sorbed to biotite and muscovite. Environ Sci Technol 38:1017–1023. https://doi.org/10.1021/es034569m
Matsuda N, Mikami S, Shimoura S, Takahashi J, Nakano M, Shimada K, Uno K, Hagiwara S, Saito K (2015) Depth profiles of radioactive cesium in soil using a scraper plate over a wide area surrounding the Fukushima Daiichi nuclear power plant, Japan. J Environ Radioact 139:427–434. https://doi.org/10.1016/j.jenvrad.2014.10.001
Kurikami H, Malins A, Takeishi M, Saito K, Iijima K (2017) Coupling the advection-dispersion equation with fully kinetic reversible/irreversible sorption terms to model radiocesium soil profiles in Fukushima Prefecture. J Environ Radioact 171:99–109. https://doi.org/10.1016/j.jenvrad.2017.01.026
http://www.tepco.co.jp/cc/press/11050804-j,/11080408-j,/11090304-j,/11100609-j,/11110406-j, and/11120810-j.html in Japanese. Accessed 17 June 2020
Sawhney BL (1972) Selective sorption and fixation of cations by clay minerals: a review. Clays Clay Miner 20:93–100. https://doi.org/10.1346/CCMN.1972.0200208
Okumura M, Kerisit S, Bourg IC, Lammers LN, Ikeda T, Sassi M, Rosso KM, Machida M (2018) Radiocesium interaction with clay minerals: theory and simulation advances Post-Fukushima. J Environ Radioact 189:135–145. https://doi.org/10.1016/j.jenvrad.2018.03.011
Strezov A, Todorova Z, Mateeva M (2000) Study of cesium and cobalt sorption and diffusion in the geological formation from Novi Han Radioactive Waste Repository, Intl workshop on experience in the upgrading of Novi Han Radioactive Waste Repository Borovets (Bulgaria) IAEA TC project BUL/4/005:9p
Mollah AS, Ullah SM (1998) Determination of distribution coefficient of 137Cs and 90Sr in soil from AERE, Savar. Waste Manag 18:287–291. https://doi.org/10.1016/S0956-053X(98)00031-2
Los Alamos Scientific Lab, NM (USA) (1980). Laboratory studies of radionuclide distributions between selected groundwaters and geologic media (LA-UR-80-437). Erdal BR. (Ed.). United States
Konishi M, Yamamoto K, Yanagi T, Okajima Y (1988) Sorption behavior of cesium, strontium and americium ions on clay materials. J Nucl Sci Technol 25:929–933. https://doi.org/10.1080/18811248.1988.9735950
Zaunbrecher LK, Cygan RT, Elliott WC (2015) Molecular models of cesium and rubidium adsorption on weathered micaceous minerals. J Phys Chem A 119:5691–5700. https://doi.org/10.1021/jp512824k
Zachara JM, Cowan CE, Resch CT (1991) Sorption of divalent metals on calcite. Geochim Cosmochim Acta 55:1549–1562. https://doi.org/10.1016/0016-7037(91)90127-Q
Bracco JN, Lee SS, Stubbs JE, Eng PJ, Jindra S, Warren DM, Kommu A, Fenter P, Kubicki JD, Stack AG (2019) Simultaneous adsorption and incorporation of Sr2+ at the barite (001) –water interface. J Phys Chem C 123:1194–1207. https://doi.org/10.1021/acs.jpcc.8b08848
Atun G, Kaplan Z (1996) Influences of salt concentration, loading and pH on strontium adsorption. J Radioanal Nucl Chem 211:425–434. https://doi.org/10.1007/BF02039708
Mimura H, Akiba K (1993) Adsorption behavior of cesium and strontium on synthetic Zeolite-P. J Nucl Sci Technol 30:436–443. https://doi.org/10.1080/18811248.1993.9734500
Carroll SA, Roberts SK, Criscenti LJ, O’Day PA (2008) Surface complexation model for strontium sorption to amorphous silica and goethite. Geochem Trans 9:2. https://doi.org/10.1186/1467-4866-9-2
Sahoo SK, Kavasi N, Sorimachi A, Arae H, Tokonami S, Mietelski JW, Łokas E, Yoshida S (2016) Strontium-90 activity concentration in soil samples from the exclusion zone of the Fukushima daiichi nuclear power plant. Sci Rep 6:23925. https://doi.org/10.1038/srep23925
Nishihara K, Iwamoto H, Suyama K (2012) Estimation of fuel compositions in Fukushima–Daiichi Nuclear Power Plant. JAEA-Data/Code 2012-018 (in Japanese) 190 pp. https://doi.org/10.11484/jaea-data-code-2012-018
Acknowledgements
This research was supported by the MEXT Nuclear Energy S&T and Human Resource Development Project through Concentrating Wisdom Grant Number JPMX15D15664655.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Sasaki, T., Matoba, D., Dohi, T. et al. Vertical distribution of 90Sr and 137Cs in soils near the Fukushima Daiichi nuclear power station. J Radioanal Nucl Chem 326, 303–314 (2020). https://doi.org/10.1007/s10967-020-07294-3
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-020-07294-3