
Abstract
Due to successes in the development on new and powerful radiopharmaceuticals, in particular of tracers for positron emission tomography, the production of a continuously expanding spectrum of radiopharmaceuticals has become more important, even for small nuclear medicine facilities. This short review summarizes and briefly describes typically established radioanalytical routines in the quality control of radiopharmaceuticals as well as the corresponding fundamental legal documents and guidelines.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The application of diagnostic radiopharmaceuticals in modern nuclear medicine can be defined as the use of radioactive probes with the aim to localize a disease and to characterize its extent and its kinetics. Ideally, this is achieved by using a targeted probe that binds with high affinity and selectivity to an extracellular or intracellular molecular target, e.g., an enzyme, a receptor, an antigen etc., whose (over)expression correlates with the extent of the disease. For positron emission tomography (PET), short lived positron emitters are nowadays routinely produced in more and more powerful compact cyclotrons by proton irradiation of small liquid or gas targets with protons [1, 2]. Apart from the frequently used cyclotron-produced PET-radionuclides 18F, 11C, 15O and 13N, that are mostly converted to the desired radiopharmaceuticals in two-to-multistep automated procedures, positron-emitting radiometals have become increasingly important [3–6]. These radionuclides, i.e., 68Ga, 64Cu and 89Zr, can be rapidly complexed by chelator-conjugated vectors (small peptides or proteins) within a few minutes in aqueous buffer in nearly quantitative yields. Furthermore, the same chelator-vector conjugates (such as DOTA peptides) are also often used for labeling with therapeutic M3+-radionuclides such as 90Y, 177Lu or 213Bi etc. During the last decade, this so called theranostic approach [7–9] that bridges the use of the same bioactive vector for a diagnostic application with the therapeutic option, has become very popular.
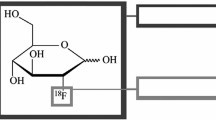
The prototype of a targeted ‘bioactive’ probe is 2-[18F]fluoro-2-deoxy-glucose ([18F]FDG), an analogue of glucose, that is transported via glucose transporters into the cell, subsequently phosphorylated by hexokinase (so called metabolic trapping) [10, 11]. [18F]FDG is widely regarded as the “blockbuster” of PET [12], and more than 116.000 publications are listed under the keyword “FDG” in Thomson Reuters “Web of Science” [13]. [18F]FDG also exemplifies the fact that targeting and detection of the rate limiting step (often the first step) of a biochemical process by a suitably designed radiotracer provides valuable information on the regulation of the entire pathway.
In order to avoid disturbance of the biochemical process of interest, radiopharmaceuticals are generally produced in high (or highest possible) specific activities. Obviously, ‘contaminations’ of a ready-to-use radiopharmaceutical formulation with labeled or unlabeled species can compete with the binding of the radiopharmaceutical and thus can result in decreased signal intensity. Determination of the chemical purity and the consideration of the presence of unlabeled (but target-binding) species is of utmost importance for the calculation of the specific activity. In the case of [18F]FDG, the presence of higher amounts of d-glucose and non-radioactive [19F]FDG (≤0.5 mg in the entire volume, e.g., FDG-TUM: ≤33.3 µg/ml; d-glucose: ≤500 µg/ml) does not alter the PET imaging results, as both rate limiting steps of the accumulation mechanism (transporter and enzyme) are only saturable in the presence of much higher amounts. In contrast, for receptor targeted probes, and in particular for small animal imaging applications in mouse models, where the human situation is reduced by a factor of 1/3000–1/4000 (body weight ratio), unsuitable specific activities or contaminations can significantly reduce the specific accumulation in the target tissue (in vivo competition effect) [14, 15]. Apart from the specific activity and the chemical purity, the radiochemical purity (presence of other labelled species) is one of the most relevant quality control parameters of radiopharmaceutical preparations. Radiochemical contaminations can result from incomplete final radio-HPLC purification (e.g., radiolabeled side products etc.), the use of impure radioisotopes (e.g., 177mLu from 177Lu as therapeutic isotope) [16], impurities from radionuclide generators (e.g., breakthrough of the 68Ge mother nuclide from 68Ge/68Ga-generators) [17], residual solvent contaminations (e.g., acetonitrile in 18F-radiopharmaceutical production from incomplete evaporation steps), or the presence of free radioisotopes (e.g., free radiometal in radio-chelate-based radiopharmaceuticals), to mention only a few. Finally, the unambiguous identification of the radionuclide incorporated into a radiopharmaceutical by means of γ-spectrometry, and, when necessary, the additional determination of the half-life (e.g., to discern one PET isotope from another), the confirmation of sterility, the absence of pyrogens, the pH, the visual aspect and the radioactive concentration are important quality control and release criteria in routine clinical radiopharmacies. Alongside with the suitable environment for radiopharmaceutical production under good manufacturing practice (GMP) conditions, this short review focuses on the state of the art radioanalytical methods and radiopharmaceutical aspects in modern clinical radiopharmacies. The review summarizes part of a presentation given at the first International Conference of Radioanalytical and Nuclear Chemistry in Budapest, May 2016.
Good manufacturing practice (GMP) and legislation
In addition to [18F]FDG with almost full market availability and some other PET tracers with marketing authorization in only a few European countries, human application of radiopharmaceuticals in clinical practice (apart from clinical studies) is often based on exemptions from the need for a market authorization as stated in Directive 2001/83, Title II [18]. Here, the permission for ‘extemporaneous’ preparation of medicinal products such as the radiopharmaceuticals used is given when a medical prescription for an individual patient has been issued (magistral preparation or approach). Great variation, however, exists concerning the interpretation of this directive within Europe. The production of radiopharmaceuticals has to meet the European GMP requirements described in vol. 4 of the Eudralex (Guidelines for good manufacturing practices for medicinal products for human and veterinary use) [19] (Table 1). This web-based compendium summarizes the general rules in vol. 4, part 1–3 and gives specific information on dedicated topics in Annexes, i.e., in Annex 3 (manufacture of radiopharmaceuticals) and Annex 13 (manufacture of investigational medicinal products). Further requirements on the automated production and subsequent instrumental analysis prior to release are covered in Annex 11 (computerized systems) and Annex 15 (qualification and validation). Since these rules and guidelines do not distinguish between the production of a few millilitre of a formulated radiopharmaceutical for a single or only a few patients and the production of several 100 kg of a drug by the pharmaceutical industry, PET centers are struggling to comply with the increasing demands and frequently changing GMP rules.
The EDQM is the organ of the European Council responsible for the European Pharmacopeia, which provides the quality standards for pharmaceutical and radiopharmaceutical preparations [20]. Over the years, several monographs on radiopharmaceuticals for PET, SPECT and endoradiotherapy have been released and published [21] (see Tables 1, 2). These monographs address specific characteristics of radiopharmaceuticals labeled with short-lived radionuclides as well as corresponding quality control procedures. Similar guidelines and documents have been published by the United States Federal Drug Administration (FDA) [22–24]. A more detailed discussion of the regulations and legislation of radiopharmaceuticals for human use is given by Decristoforo and Schwarz [25].
Typical quality control procedures in a clinical radiopharmacy
For quality control (QC) purposes (see Table 2), three samples of the final product are typically taken under laminar flow or sterile conditions (class A): (a) approx. 500 µl for sterility tests, (b) approx. 500 µl for general quality control purposes (see below) and (c) approx. 1 ml as a reference sample (retained sample which is stored for at least one year).
Sterility and endotoxin testing
One example of a QC criterion specific for short-lived radiopharmaceuticals is the allowance to release these radiopharmaceuticals before completion of all test, in particular of tests (such as sterility tests) that are not compatible with the short half live. In these cases, sterilization of the final product or final sterile filtration (0.22 µm filter) plus performance of a filter integrity test (e.g., bubble point test) prior to release is required. Former time-consuming endotoxin tests have been substituted by modern and rapid automated endotoxin testers that are able to provide valid results within a few minutes.
Appearance
The ‘appearance’ test is a general test to be carried out on all products (generally: liquids ready for injection with a volume of 8 to ca.15 mL) to confirm the absence of visible particles, turbidity or color. For this purpose, the vial is inspected by means of tweezers through a lead window of a typical U shape shielding where the samples taken for quality control are placed (in the QC lab) during the QC procedure.
Identity, chemical purity and radiochemical purity
Radio-HPLC is probably the most important and most often used QC-procedure. With radio-HPLC, several fundamental characteristics can be determined (although in some cases not completely): (a) the incorporation of the radioisotope into the correct molecular species (the ‘identity’ of the radiopharmaceutical, e.g., radiometallated peptides or antibodies, 18F-labelled small molecules etc.), (b) the presence of other radiolabelled species that do not co-elute with the product; e.g., other small molecules labelled with 18F, protein fragments or oxidized radiometallated peptide species formed by radiolysis during the production etc. (radiochemical purity), and (c) the presence of non-labelled organic contaminations, e.g., residual precursor, fragments, elimination products, residual reagents etc. (chemical purity). Obviously, a broad range of HPLC columns is necessary to be able to analyze radiopharmaceuticals with highly variable physicochemical characteristics: (a) RP18 reversed phase columns for almost all 18F- and 11C-compounds, (b) anion exchange chromatography for e.g., the analysis of 18F-FDG or 18F-fluoride solutions (or cation exchange chromatography and ion pair chromatography for others), or (c) size exclusion chromatography for proteins such as radioiodinated albumin or proteins. In combination with these columns, either UV detectors, electrochemical or refractive index detectors (for FDG) are commonly used. However, some data cannot be acquired by radio-HPLC. Since an HPLC profile only reflects the mass profile of compounds eluted from the column during a dedicated time span, it is often unclear whether additional compounds that are retained more strongly on the column or even adhere to the column material are not susceptible of quantitative analysis or converted during the HPLC analysis. A prominent example it the radio-HPLC analysis [18F]FDG, where non-hydrolyzed (still acetyl protected) tetra-acetyl-2[18F]glucose or partly hydrolyzed products are never detected, even when they are present in the final product: since the mobile phase used for the strong basic anion-exchange chromatography of small carbohydrates is 0.1 M NaOH, the protecting groups (acetyl groups) are cleaved during the radio-HPLC analysis of [18F]FDG thus not allowing to quantify the presence of only partly hydrolyzed product. In this context and as a general second chromatography method, radio-TLC instruments are commonly installed in modern radiopharmacies.
For 99mTc kit preparations, only approved 99mTc generators and approved lyophilized kits are used. For such kit preparations, radio-TLC (or paper chromatography) represents the primary method for quality control, besides being used as a second additional chromatographic analysis for other radiopharmaceuticals. A variety of commonly used mobile phases that guarantee optimal separation of the labeled product and radioactive contaminations and thus provide valid QC results have been investigated and published.
Radionuclide identity and radionuclide purity (radionuclidic impurities)
Radionuclide identity and radionuclide purity (radionuclidic impurities) are investigated by means of γ-spectrometry. Whereas the determination of the characteristic γ-emissions clearly indicates the presence of most of the SPECT (123I, 111In, 99mTc etc.) and therapeutic radioisotopes (177Lu, 131I, 90Y etc.), PET isotopes can be identified as such by the presence of a 511 keV (and a sum peak at 1022 keV), but cannot be distinguished from one another. Thus, determination of the half-life over a period of at least three half-lives of the respective radionuclide of interest need to be carried out, typically by linear regression of data obtained with a γ-spectrometer or a sufficiently sensitive dose calibrator. In some cases it is not necessary to carry out this test for all production runs. Thus, for example, only one initial determination of the half life of 18F-fluoride produced by irradiation of a new batch of [18O]H2O is required. Similarly, the identity and purity of 68Ga3+ (T 1/2 = 68 min) eluted from a 68Ge/68Ga-generator is investigated approx. once a month. For this purpose, the γ-spectrum and the decay of a defined sample of the generator eluate is measured immediately after elution (68Ga) and 48 h after elution (on the same sample: 68Ge-breakthrough) [18]. Another important example is the presence of up to 0.4 kBq 177mLu (T 1/2 = 161 days) per MBq 177Lu (T 1/2 = 6.7 days) at the end of neutron irradiation when 177Lu is produced by the 176Lu (n, γ) 177Lu process, since these long lived 177mLu contaminations can cause significant problems when mixed with other typically used therapeutic radionuclides, such as 131I, in decay tanks of hospitals [16]. In vivo problems (dosimetry) with small 177mLu contaminations in 177Lu-radiopharmaceuticals have not been reported so far.
Volume, radioactivity concentration, shelf life
The overall volume of a final radiopharmaceutical ready for injection is also important. In accordance with national laws, radiopharmaceuticals are treated as drugs; thus the effect (the PET image quality) of the medication (the PET radiopharmaceutical) must be ensured until the end of the shelf life. Consequently, the shelf life is defined as the time point when the radioactivity of the entire product volume with the minimum specified activity concentration (at the end of production) has to be injected into one patient to obtain a PET image of suitable quality. In addition, it has to be demonstrated that the product meets all specifications, i.e., the criteria defined for the radiochemical purity (affected by radiolysis over time) at the end of the shelf life. If radiolysis is fast, the shelf life is determined by the radiochemical purity. As the kinetics of radiolysis have to be investigated during the validation process of each radiopharmaceutical, the maximum radioactivity concentration (e.g., GBq/ml) is determined by the highest activity concentration tested for radiolysis in these validation runs.
Residual solvents
Since the maximum volume (typically between about 8–15 ml) also determines the maximum injected dose of e.g., residual solvents, such as ethanol from solid phase extraction procedures (e.g., FDG-TUM: 1200–2500 µg/ml; to ensure radiolytic stability, lower limit must be fulfilled; see Table 3) or acetonitrile (limit in FDGTUM: 270 µg/ml) from 18F-fluorinations etc. to be injected into a patient at the end of the shelf life, maximum doses (e.g., in mg/V; V entire volume) are also specified for typical impurities (see Table 1). As demonstrated by the use of V for the specification of the maximum injectable dose for each impurity, the overall volume must also be reproducible and within a well-defined and suitably narrow range.
pH and osmolality
The determination of the pH is also routinely carried out. Although not mentioned in the specific monographs, the determination of the osmolality (human plasma osmolality: 290 m Osm/l, range: 285–310 mOsm/l; former limit for FDG-TUM: 350–550 mOsm/kg) is also carried out by some radiopharmacies.
As a typical example, Table 3 summarizes the release criteria of FDG-TUM (FDG with market authorization produced at the Technische University Munich). Apart from the methods, two columns (old/new specifications) are listed. The ‘old’ specifications were established in 2000 as part of the approval process for market authorization. At that time, FDG was produced from [18F]fluoride generated by means of a RDS cyclotron, and the synthesis was carried out by a Siemens CTI CPCU-module. To meet the continuously increasing GMP demands and to be able to use a new cyclotron (GE PETtrace 800; installed in 2014) and a new FASTlab-module for the approved FDG-TUM production process, new validation runs were carried out and new specifications and release criteria were compiled. The latter was accomplished in close interaction with the local authorities.
As demonstrated in Table 3, the osmolality test was omitted, and some specifications were modified (e.g., determination of fluorodeoxy-mannose (FDM) instead of chlorodeoxy-glucose (ClDG), since in the new process HCl deprotection of the tetra-acetylated mannose triflate precursor was substituted by alkaline hydrolysis). In addition, frequencies of tests (e.g., radionuclide identity and radionuclide impurities only once per month and once for each new [18O]H2O batch) or acceptable ranges (pH) etc. were changed, demonstrating that GMP (or cGMP; current Good Manufacturing Practice, as used in the US) is not an inflexible and rigid system of guidelines, but a dynamic and changing system that is continuously adapted to the current state of knowledge and experiences.
Outlook
After a decade of outstanding technical achievements in molecular imaging, this expertise is now expected to add more momentum and innovation to the advancement of radiopharmaceuticals for molecular imaging and radionuclide therapy. However, this innovation pressure is accompanied by continuously rising demands and legal requirements on clinical GMP production of radiopharmaceuticals. It will be of utmost importance to establish a new research focus that allows to suitably address this topic of continuously growing importance. In view of the cost explosion in that area, appropriate and sustainable strategies must be defined, and solutions need to be developed and elaborated. Without such perspectives and solutions, the transfer of new diagnostic and therapeutic radiopharmaceuticals into clinical application—the final objective of translational radiopharmaceutical development—will in the future only be possible with considerable financial expenditure.
References
Weissleder R, Pittet MJ (2008) Imaging in the era of molecular oncology. Nature 452(7187):580–589
Wester HJ (2007) Nuclear imaging probes: from bench to bedside. Clin Cancer Res 13(12):3470–3481
Jacobson O, Kiesewetter DO, Chen X (2015) Fluorine-18 radiochemistry, labeling strategies and synthetic routes. Bioconjug Chem 26(1):1–18
Braghirolli AM, Waissmann W, da Silva JB, dos Santos GR (2014) Production of Iodine-124 and its applications in nuclear medicine. Appl Radiat Isot 90:138–148
Velikyan I (2015) 68Ga-based radiopharmaceuticals: production and application relationship. Molecules 20(7):12913–12943
Kasbollah A, Eu P, Cowell S, Deb P (2013) Review on production of 89Zr in a medical cyclotron for PET radiopharmaceuticals. J Nucl Med Technol 41(1):35–41
Park JA, Kim JY (2013) Recent advances in radiopharmaceutical application of matched-pair radiometals. Curr Top Med Chem 13(4):458–469
Schottelius M, Wirtz M, Eiber M, Maurer T, Wester HJ (2015) [111In]PSMA-I&T: expanding the spectrum of PSMA-I&T applications towards SPECT and radioguided surgery. EJNMMI Res 5(1):68
Srivastava SC (2012) Paving the way to personalized medicine: production of some promising theragnostic radionuclides at Brookhaven National Laboratory. Semin Nucl Med 42(3):151–163. doi:10.1053/j.semnuclmed.2011.12.004
Greenberg JH, Reivich M, Alavi A, Hand P, Rosenquist A, Rintelmann W, Stein A, Tusa R, Dann R, Christman D, Fowler J, MacGregor B, Wolf A (1981) Metabolic mapping of functional activity in human subjects with the [18F]fluorodeoxyglucose technique. Science 212:678–680
Reivich M, Kuhl D, Wolf A, Greenberg J, Phelps M, Ido T, Casella V, Fowler J, Hoffman E, Alavi A, Som P, Sokoloff L (1979) The [18F]fluorodeoxyglucose method for the measurement of local cerebral glucose utilization in man. Circ Res 44(1):127–137
Fowler JS, Ido T (2002) Initial and subsequent approach for the synthesis of 18FDG. Semin Nucl Med 32(1):6–12 (Review)
Notni J, Steiger K, Hoffmann F, Reich D, Kessler H, Schwaiger M, Wester HJ (2016) Variation of specific activities of Ga-68-aquibeprin and Ga-68-avebetrin enables selective PET-imaging of different expression levels of integrins α5β1 and αvβ3. J Nucl Med 57(10):1618–1624
Velikyan I, Sundin A, Eriksson B, Lundqvist H, Sörensen J, Bergström M, Långström B (2010) In vivo binding of [68Ga]-DOTATOC to somatostatin receptors in neuroendocrine tumours–impact of peptide mass. Nucl Med Biol 37(3):265–275
Bakker WH, Breeman WA, Kwekkeboom DJ, De Jong LC, Krenning EP (2006) Practical aspects of peptide receptor radionuclide therapy with [177Lu][DOTA0, Tyr3]octreotate. Q J Nucl Med Mol Imaging 50(4):265–271
de Blois E, Sze Chan H, Naidoo C, Prince D, Krenning EP, Breeman WA (2011) Characteristics of SnO2-based 68Ge/68Ga generator and aspects of radiolabelling DOTA-peptides. Appl Radiat Isot 69(2):308–315
The European Parliament and the Council of the European Union (2001) Directive 2001/83/EC of the European Parliament and the Council of 6 November 2001 on the community code relating to medicinal products for human use. Off J Eur Union 311:1–66
EU Pharmaceutical Information (2003) EudraLex—volume 4 good manufacturing practice (GMP) guidelines. http://ec.europa.eu/health/documents/eudralex/vol-4/index_en.htm
European Directorate of Quality of Medicines (2011) Compounding of radiopharmaceuticals. Pharmeuropa 23(4):643
European Pharmacopoeia (Ph. Eur.) Vol 8 (2013–2016) European Directorate of Quality of Medicines
US Food and Drug Administration (2011) Federal register. http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=212
US Food and Druig Administration (2011) Guidance PET drugs—current good manufacturing practice (CGMP), small entity compliance guide. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM266640.pdf
United States Pharmacopeia (2010)
Decristoforo C, Schwarz SW (2011) Radiopharmacy: regulations and legislations in relation to human applications. Drug Discov Today Technol 8(2–4):e71–e77
Acknowledgements
Funding was provided by Deutsche Forschungsgemeinschaft (Grant No. SFB824, subproject Z1).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Schmidt, A., Schottelius, M., Herz, M. et al. Production of clinical radiopharmaceuticals: general pharmaceutical and radioanalytical aspects. J Radioanal Nucl Chem 311, 1551–1557 (2017). https://doi.org/10.1007/s10967-016-5125-6
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-016-5125-6