
Abstract
The determination of 236U with accelerator mass spectrometry (AMS) requires efficient separation methods with high concentration factors. The article proposes an alternative method of uranium separation from aqueous solutions to commonly used iron co-precipitation. This “homogeneous precipitation” is based on unique properties of fresh hydrous titanium oxide, prepared by hydrolysis of its organic precursor—tetra-n-butylorthotitanate—directly in the aqueous sample solution. Besides high uranium uptake, the results shows very promising properties of this uranium-titanium oxide precipitate for 236U-AMS measurements providing up to 4 times higher negative ion yield comparing with common uranium oxide matrix.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In environmental studies the determination of low concentrations has always been a key issue and therefore, the concentration step is usually a crucial part of the sample processing. For separation of uranium, various pre-concentration techniques have been developed and critically reviewed by Rao et al. [1]. The extensive efforts have been dedicated to the use of efficient sorption materials, both organic and inorganic [2]. Among these, the inorganic materials based on titanium dioxides have been extensively studied and have shown promising results [3, 4].
For ultra-low concentration measurements, special detection techniques are still highly needed, despite the usage of efficient sorption materials in the sample preparation procedures. Accelerator Mass Spectrometry (AMS) has proven to be a suitable detection technique with high sensitivity, selectivity and low detection limits [5].
In our previous work, various types of titanium dioxide prepared by the hydrolysis of tetra-n-butylorthotitanate were characterized including their sorption properties towards uranium from which one of the materials showed the sorption capacity higher than 260 mg uranium per gram of dry titanium dioxide [6].
This research is now focused on the utilization of titanium dioxide (TiO2) properties as a potential AMS target matrix, and in parallel also as a sorption material in the precipitation of titanium dioxide by the hydrolysis of tetra-n-butylorthotitanate with simultaneous uranium sorption. This “homogeneous precipitation” was already successfully used for the production of uranium doped TiO2 nanomaterials and advantages like short synthesis time and simple operation were highlighted [7]. The implementation of titanium dioxide materials as a sorption material and as a target matrix into classic methods of sample preparations for 236U/238U measurements using AMS would mean a significant reduction of separation steps in procedures and simplification of the whole method.
Experimental
Reagents and chemicals
In the basic studies of homogeneous precipitation, tetra-n-butylorthotitanate (TBOT, C16H36O4Ti; Sigma-Aldrich) and uranyl nitrate of p. a. grade (Lachema) were used. Since commercial uranyl salts may contain high 236U/238U isotopic ratio, uranium from the Vienna-KkU in-house standard with the 236U/238U isotopic ratio of (6.98 ± 0.32) × 10−11 was used for the AMS measurements [8].
The spectrophotometric measurements were carried out with arsenazo III and hydrochloric acid of p.a. quality and measured using Helios ε (ThermoSpectronic).
For liquid scintillation measurements, Rotiszint Eco, resp. Ultima Gold AB (Carl Roth GmbH & Co., resp. Perkin Elmer) scintillation cocktails and 6 or 20 mL plastic vials (P–Lab) were used.
“Homogeneous precipitation”
Tetra-n-butylorthotitanate (100, 50 and 20 µL) was added dropwise into the uranium solutions (0.5, 2 and 20 mmol L−1) while mixing on the magnetic laboratory stirrer. The initial pH values of the solutions were 3.88, 3.40 and 2.43, respectively. The suspension was mixed for 2 h and afterwards, filtered using 0.45 µm filters (MFTM Membrane filters HA, Merck). The aliquots of the filtrate and the original solution were taken for uranium concentration measurements. The solid residue was put into an ampoule together with the filter and was dried at 30 °C in the vacuum dryer (p ~0.5 kPa) before determining the weight.
From the activity of solutions before (A 0) and after (A) precipitation, relative uptake u was calculated and used for the evaluation.
Uranium measurements
For the highest uranium concentration in the microgram range, the spectrophotometric determination with arsenazo III was used and the evaluation was performed at a broad band with maximum absorbance at 651 nm. The solutions of the samples were prepared in the following way: 3 mL of 0.2 mM arsenazo III solution and 5 mL of the samples were put into a volumetric flask, then filled up to 25 mL with hydrochloric acid and water to reach 0.01 M acidity [9].
The uranium solutions of milligram range concentration were measured using Liquid Scintillation counting (LSC; Triathler Hidex Oy or Hidex 300 SL, Finland). Aliquots were mixed in vials with scintillation cocktails and the settings (α window, PLI, time etc.) were chosen based on the matrix of the sample. The measuring time and the amount of the sample were adjusted in order to reach sufficient count rates (0.1–40 CPS).
Matrix preparation
The newly studied matrix based on titanium dioxide was compared with the classical oxide one which was used as a reference and produced by combusting Vienna-KkU uranyl nitrate at 900 °C for 4 h. The samples made of titanium dioxide and known uranium amount were prepared and to avoid possible anthropogenic contamination, Vienna-KkU uranyl nitrate was used. Into 10 mL of uranium solution (9.46 mg of Vienna-KkU in 10 mL) 100 µL of TBOT was added dropwise and the suspension was stirred for 1 h. The suspension was filtered on 0.2 µm filter paper (NC20 Schleicher&Schuell MicroScience GmbH) using a water jet pump. The filter paper together with the residue was placed into a crucible and combusted at 900 °C. Aliquots of the solutions before and after precipitation were taken for the alpha measurements using LSC. The sample was pressed into aluminium sample holders suitable for the ion source of the AMS.
AMS measurements
All measurements were carried out at the VERA facility at the University of Vienna, Austria [10]. The details of the measurement procedure are following. The samples are sputtered with Cs+ ions at the energy of 5 keV and only a small fraction of the uranium atoms are converted to the proper ion species UO− for the AMS instrument: for UO− the ion yield from the classical iron oxide matrix is about 0.45 % [11]. After pre-acceleration to 55 keV, the negative ions undergo a first mass separation in electric and magnetic sectors fields (Δm/m ≈ 500) and are accelerated to 3 MeV in the first stage of a tandem accelerator. At the high–voltage terminal, the particles interact in the so-called stripper tube (1 m length, about 0.3 Pa) with Ar gas, which breaks up the molecules and converts about 5 % of the actinides into the 5+ charge state. The positive ions including the molecular fragments are again accelerated in the second stage of the tandem accelerator, producing U5+ at 18 MeV. A series of magnetic and electric sectors purifies the beam, down to an abundance sensitivity of at least 10−12 [8]. The ions are finally detected by an energy-dispersive ionization detector.
The performance and background of the system depends strongly on the sample matrix used. Thus, the full negative ion spectra were analysed from 10 to 270 amu extracted from both a U + Ti mixture and a pure uranium oxide sample. Additionally, the material in three sample holders with titanium oxide matrix, and three sample holders with pure uranium oxide were completely sputtered away. In this experiment, pure uranium oxide was used for comparison, despite mixing with iron powder (or e.g. silver used at other labs) is known to increase the negative ion yield; however, this improvement depends on the precise mixing ratio, and probably also grain size and homogeneity of the mixing. The yield of pure uranium oxide is probably better reproducible at other labs and is thus better suited as a reference.
To reduce the effort involved in the ionization yield measurements presented, a maintenance shut-down of the tandem accelerator was used, where the otherwise idle injector was operated independently for about 3 days, and only negative ions 238UO− were measured. Only during about one work day (10 h), the beam was injected into the accelerator, and 238U5+ and 236U3+ were detected.
Results and discussion
Uranium sorption
The successful preparation of new hydrous titanium oxide with various sorption properties published in our previous paper [6] was the origin of the idea to join the hydrolytic preparation with the sorption properties of fresh precipitate into the so called “homogeneous precipitation” and to propose it as a possible alternative separation method which can minimize the number of steps in preparation procedures and which may partially or even fully replace the iron hydroxide co-precipitation used in almost each 236U sample preparation procedure [12, 13]. The main disadvantages of this common coprecipitation are the use of commercial iron compounds, which may contain anthropogenic contamination and thus negatively influence the 236U/238U isotopic ratio, and the use of the very same iron hydroxide in all of the subsequent multiple dissolutions of the whole procedure.
As a result of the hydrolysis reaction between TBOT and water, hydrous titanium oxide is formed and immediately during its formation sorbs or co-precipitates ions from the solution. The performed experiments showed consistent results and the mass of the formed solid material is proportional to the added volume of TBOT with the slope of mass (mg) to volume (μL) dependency of 0.319 ± 0.004 (R2 = 0.978, set of 29 experiments); hence, the procedure gives repeatable results for the purposes of the experiment. The value of the slope agrees with the fact that approximately two-thirds of the TBOT mass comes from aliphatic chains and only the last third part forms the mass of hydrous titanium oxide.
The sorption of uranium from aqueous solutions is also proportional to the added TBOT amount and it decreases with increasing uranium concentration (Fig. 1). Solutions before and after the precipitation were checked for the change in pH. With increasing uranium concentration, pH after precipitation slightly decreases by up to 0.6 units in the case of 0.5 mM uranium solutions.

The effect of TBOT volume (50, 100 and 200 μL) added into uranium solution of various concentrations (20, 2, 0.5 mmol L−1) stirred for 2 h (14 mL of solution) with eyeguides
For studying the adsorption, the obtained data were used to determine the isotherm type. The linear regression of the Langmuir type can be recognised in the relation between 1/q and 1/c (q, c—concentrations of species in the solid and liquid phase, respectively), whereas for the Freundlich type in the relation between logq and logc. The best fits for the adsorption of uranyl ions are given for the Freundlich isotherm and are shown in Fig. 2.

Freundlich and Langmuir isotherm plot for uranium sorption on fresh hydrous titanium oxide during “homogeneous precipitation”—equilibrium concentrations in solution c and on absorber q in relation with the added amount of 200 μL TBOT (initial uranium concentration: 0.5, 5 and 20 mmol L−1; 2 h of mixing)
The applicability of this separation method is also supported by the promising results obtained in the experiment with a real water sample from the inlet to the mine water treatment plant placed in Dubenec, Bytíz, CZ (CDV PB II). This water sample was not acidified and after spontaneous iron precipitation, the uranium concentration in the clear solution was externally determined to be 5.29 mg L−1. The experiment was performed with 750 mL water sample into which only 1 mL of TBOT was added dropwise. The uranium uptake of 56 % was calculated from the uranium concentration before and after the “homogeneous precipitation” measured by the spectrophotometric method with arsenazo III.
The feasibility of “homogeneous precipitation” method was shown on the treatment of the real water sample and regarding the requirements for the determination of 236U/238U isotopic ratio, this yield of uranium from such mineralised water is sufficient and promising for also other possible applications. However, to understand this process more experiments have to be carried out.
AMS measurements
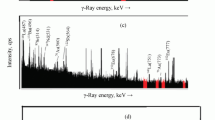
Firstly, mass scans of both matrix samples were measured and the comparison in the measured masses can be seen in Fig. 3. Starting from the lightest side, the oxide peaks (mass 16 and mass 32) are strong from both sample types and the next peaks can be attributed to the ions containing 27Al (27Al−, 27Al16O−, Al2 −, AlO2 −, Al4 −···) originating from the sample holder. The higher current at mass 28 in the titanium oxide matrix may be 27AlH−, an indication for a higher hydrogen content in this matrix, but this would also result in higher OH− current which was not observed. Hydride ions might cause a problem for 236U measurements, as 235UOH− is the limiting factor in conventional mass spectrometry, and also involved in most background processes in AMS [14]. However, the peak at mass 28 can be also explained as 28Si−, with matching peaks for 29Si− and 30Si−. Also peaks for 28SiO− and 28SiO2 − are present. The presence of Si is unexpected from the perspective of the preparation procedure; however, as the associated peaks do not influence the 236U determination, its origin was not further investigated. Only a weak peak is visible at mass 48 (48Ti−), which reflects the low electron affinity of 0.08 eV of the atom. Prominent is the 48Ti16O−, accompanied by analogous peaks of the four minor titanium isotopes 46Ti–50Ti. This pattern repeats with decreasing intensity for TiO2 − and TiO3 −. In addition, some Ti–Al clusters also with 12C and 16O are observed (mass 99 and, weaker mass 75 and mass 123). 238U− is only weakly observed (the electron affinity is unknown), the most intense uranium anion is UO− with current for both matrices. Overall, none of the observed ion species is likely to interfere with a 236U measurement and from this point of view the U + Ti matrix is suitable for further research for its AMS applications.

Mass scans of uranium (red) and titanium oxide with uranium (blue). All larger peaks from the titanium oxide matrix could be identified (see text). (Color figure online)
A complete sputtering of a sample in the range of a few mg of material takes on the order of 10 h at VERA. This is much more than is usually available for a routine measurement, where the affordable maximum is 1–2 h. This has led to the situation that in most cases AMS samples are not used up, and that many laboratories publish extracted ion currents instead of ionization yields. These ion currents are important technical parameters; however, they depend strongly on source type and operation parameters, and are thus hardly comparable between different labs. The total negative ion yield, on the other hand, seems to be a property of the sputter source principle, sample matrix, and ion species, which is reproducible between different facilities.
Figure 4 shows the current trend while sputtering away the available material of three pure uranium oxide and three U + Ti oxide samples. The sample changer switched between the six targets about every 30 min, total sputter duration was more than 10 h on each sample. The pure uranium oxide samples contain 0.93–2.14 mg U, while the titanium oxide matrix carries only 0.13–0.25 mg U. By integrating the current yields, the total negative ion yield can be calculated and is shown in Table 1 reaching the values of 0.16–0.23 and 0.73–0.88 % for the uranium oxide and U + Ti samples, respectively. In addition, one target of U + Ti was not exhausted even after 10 h. Even when we consider that the UO− yield of uranium oxide sample could probably be increased to 0.45 % by mixing with iron powder, U + Ti seems advantageous by a significant margin. The observed differences could be explained by the fact that during the co-precipitation uranium can get into the crystal lattice of newly-formed hydrous titanium oxide. This is also supported by the results published by Raindl who found out that uranium separated from the solution by the “homogeneous precipitation” method cannot be quantitatively eluted without a complete dissolution of titanium dioxide [15]. This observation together with higher chemical stability of TiO2 compared to uranium oxides—assuming that during sputtering of U + Ti samples, more stable TiO2 particles are destructed more slowly thus gradually exposing the incorporated uranium—could lead to higher ionization yield and also higher yield stability in time.

Time trend of UO− current from uranium oxide (KkU-1, 2, 3) and U-spiked TiO samples (TiOKkU-1, 2, 3). After about 2000 s of measurement on each target, the source power was reduced to achieve more stable output of the uranium oxide targets. Currents are normalized to uranium mass in the target
The AMS measurement results are also shown in Table 1. The 236U/238U isotopic ratios and average 238U5+ currents were actually calculated only from three points in Fig. 4 after almost 30,000 s of sputtering, when the tandem accelerator was available. Between 142 and 6053 236U5+ ions were detected during a net detector live time of 4370 s on each sample. Both the 238U5+ current and the 236U5+ count rate are roughly proportional to the uranium amount in the samples of each material. The uncertainty given in Table 1 includes both counting statistics and the reproducibility of the three measurements on each sample. The nominal ratio of Vienna-KkU is (6.98 ± 0.32) × 10−11 [8]. The measured isotopic ratios are in good agreement with this value within their uncertainties, but for the titanium oxide matrix with the largest uranium amount; this sample has probably an underestimated uncertainty due to the high number of 236U counts. Detection limits can be estimated relative to the discussion in [14], which is based on an assumed ionization yield of 0.3 % for the uranium oxide matrix. Our observed ionization yield of ~0.8 % from the TiO2 matrix, combined with helium stripping and an ionization chamber as particle detector, translates to a detection efficiency of 1.3 × 10−3, i.e. one out of 750 atoms in the sample could be detected. For real samples, the effective efficiency may be, however, limited by 236U background in sample preparation and by instrumental background. The latter is mainly associated with 235UOH− molecules, which should not be affected by the presence of titanium in the sputter matrix. No peaks are visible in the anion mass spectra of the TiO2 sputter targets, which could interfere with the measurement. Therefore, we expect a similar isotopic abundance limit of 2 × 10−14 (with oxygen stripping and time-of-flight detector) as given in [14], but with the possibility to use smaller samples due to the higher detection efficiency.
Conclusions
Precipitation of TiO2 via in sample hydrolysis of TBOT—here called “homogenous precipitation” allows high and reproducible yields of sorbed uranium. The procedure also provides repeatable amounts of resulting TiO2 precipitate. For the real samples, homogenous precipitation is usable for uranium separation also at very high volume-over-mass (V/m) ratios, in case of V/m = 2350 mL mg−1 uptake of 56 % of uranium was observed. This is very promising for pre-concentration of uranium from real water samples.
To our best knowledge, titanium dioxide was used as AMS target matrix for the first time. “Homogenous precipitation” results in a TiO2 matrix providing up to 4 times higher negative ion yields in the caesium sputter source than the standard uranium oxide matrix. Mass scans also show that this matrix is not contributing any molecular interferences and thus is suitable for 236U AMS measurement. The stability of the new matrix allows long and stable measurements, thus improving statistics of the final result.
Combination of high uptake of uranium and high ionization yield may result in very efficient one-step sample preparation. A four times lower volume of clear water samples compared to iron co-precipitation procedure may be treated directly with TBOT, and after filtration and combustion the precipitate may be directly pressed into AMS sample holders.
Up to now, the selectivity of “homogenous precipitation” was not tested and the uptakes of other actinides are not yet known. This could make the “one-step” procedure inappropriate/unsuitable for some applications but for others could be advantageous.
For chemical separation the additional disadvantage may be the strong uptake of uranium, probably incorporated into TiO2 matrix, and rather difficult uranium release from the precipitate back to the solution. In these cases, the most suitable application of TiO2/TBOT would be during the final steps of target preparation.
References
Rao TP, Metilda P, Gladis JM (2006) Preconcentration techniques for uranium(VI) and thorium(IV) prior to analytical determination—an overview. Talanta 68:1047–1064
Rao L (2011) Recent International R&D Activities in the Extraction of Uranium from Seawater. Lawrence Berkeley National Laboratory, Berkeley
Morrison SJ, Spangler RR (1992) Extraction of uranium and molybdenum from aqueous solutions: a survey of industrial materials for use in chemical barriers for uranium mill tailings remediation. Environ Sci Technol 26:1922–1931
Motl A, Šebesta F, John J, Ndiaye I, Němec M, Špendlíková I (2013) Comparison of uranium extraction from model fresh water on TiO-PAN and NaTiO-PAN composite absorbers. J Radioanal Nucl Chem 298:2057–2063
Hellborg R, Skog G (2008) Accelerator mass spectrometry. Mass Spectrom Rev 27:398–427
Špendlíková I, Raindl J, Němec M, Steier P, Mičolová P (2014) Preparation of pure TiO2 sorption material. J Radioanal Nucl Chem 300:1151–1158
Liu SY, Feng QG (2011) Synthesis of uranium doped TiO2 nanomaterials and its visible light degradation property. Adv Mat Res 148–149:1208–1211
Steier P, Bichler M, Fifield LK, Golser R, Kutschera W, Priller A, Quinto F, Richter S, Srncik M, Terrasi F, Wacker L, Wallner A, Wallner G, Wilcken KM, Wild EM (2008) Natural and anthropogenic 236U in environmental samples. Nucl Instrum Meth B 266:2246–2250
Rohwer H, Rheeder N, Hosten E (1997) Interaction of uranium and thorium with arsenazo III in an aqueous medium. Anal Chim Acta 341:263–268
Steier P, Dellinger F, Forstner O, Golser R, Knie K, Kutschera W, Priller A, Quinto F, Srncik M, Terrasi F, Vockenhuber C, Wallner A, Wallner G, Wild EM (2010) Analysis and application of heavy isotopes in the environment. Nucl Instrum Meth B 268:1045–1049
Fifield LK, Clacher AP, Morris K, King SJ, Cresswell RG, Day JP, Livens FR (1997) Accelerator mass spectrometry of the planetary elements. Nucl Instrum Meth B 123:400–404
Qiao J, Hou X, Steier P, Golser R (2013) Sequential injection method for rapid and simultaneous determination of 236U, 237Np, and Pu isotopes in seawater. Anal Chem 85:11026–11033
Srncik M, Steier P, Wallner G (2010) Determination of the isotopic ratio 236U/238U in Austrian water samples. Nucl Instrum Meth B 268:1146–1149
Winkler SR, Steier P, Buchriegler J, Lachner J, Pitters J, Priller A, Golser R (2015) He stripping for AMS of 236U and other actinides using a 3 MV tandem accelerator. Nucl Instrum Meth B 361:458–464
Raindl J (2013) Development and testing of procedures for uranium separation from natural waters with sorption materials based on TiO2. Diploma thesis, Prague, Czech Technical University in Prague (in Czech)
Acknowledgments
This research has been supported by the Grant Agency of the Czech Technical University in Prague, grant No. SGS 14/154/OHK4/2T/14, by the Ministry of Industry and Trade of the Czech Republic under grant No. FR-TI3/245, by the Ministry of Education, Youth and Sports of CR under grant No. 7AMB12AT022 and by the Austrian Agency for International Cooperation in Education and Research under grant No. CZ14/2012.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Špendlíková, I., Němec, M., Steier, P. et al. Sorption of uranium on freshly prepared hydrous titanium oxide and its utilization in determination of 236U using accelerator mass spectrometry. J Radioanal Nucl Chem 311, 447–453 (2017). https://doi.org/10.1007/s10967-016-5013-0
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-016-5013-0