
Abstract
Amid the global water crisis, developing innovative solutions for sustainable water purification is crucial. Membrane distillation (MD) has emerged as a pivotal method with significant potential to address these challenges effectively. In this research endeavor, our team focused on creating and optimizing an omniphobic membrane utilizing a combination of Poly(vinylidenefluoride-co-hexafluoropropylene) (PVDF-HFP), Tetraethyl orthosilicate (TEOS), and Perfluorodecyltrimethoxysilane (PFDTMS) through the nonsolvent-induced phase separation (NIPS) methodology. The resultant membrane displayed a notable contact angle of 130°, underscoring its remarkable omniphobic properties. Various characterization techniques were used to evaluate the membrane's performance, including liquid entry pressure (LEP), porosity analysis, thickness determination, pore size quantification, and scanning electron microscopy (SEM). SEM images revealed enhanced surface roughness, which is critical for efficiency. Additionally, X-ray diffraction (XRD), Fourier-transform infrared spectroscopy (FTIR), and energy-dispersive X-ray spectroscopy (EDS) provided insights into the membrane's crystalline structure and chemical composition.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Membrane distillation (MD) represents a thermally activated separation technique applicable to desalination and wastewater treatment. Unlike pressure-driven membrane methods like reverse osmosis, MD functions under low feed pressure and is minimally impacted by feed solution concentration, facilitating the realization of zero liquid discharge integrated processes. [1,2,3]. Compared to traditional thermal evaporation techniques, MD units can be tailored to optimize the surface-to-volume ratio (referred to as packing density), thereby enabling increased evaporation areas within smaller facilities [4, 5]. Furthermore, as MD typically operates at reduced temperatures (e.g., below 80 °C), it can leverage low-grade thermal resources such as solar or industrial waste heat. Despite establishing certain industrial-scale facilities in recent years, large-scale deployment of MD is impeded by technological barriers and primarily by the absence of highly efficient membranes [6,7,8,9].
In nonsolvent-induced phase separation (NIPS), a polymer solution is cast onto a flat surface and submerged in a suitable nonsolvent medium. A solvent/nonsolvent substitution occurs, and upon reaching a critical composition, the polymer solution undergoes thermodynamic instability, leading to liquid–liquid phase separation into polymer-rich and polymer-poor solutions [10,11,12].
The kinetics of phase separation are markedly influenced by the constituents of the system and their concentrations. Specifically, the nonsolvent is considered potent when its interaction with the polymer is weak, and the rate of solvent/nonsolvent exchange is rapid. Under these circumstances, instantaneous phase separation of the polymer solution occurs, resulting in a membrane characterized by a dense upper layer on a substrate featuring finger-like or pear-shaped macrovoids [13, 14]. Conversely, if the nonsolvent exhibits a stronger affinity towards the polymer, precipitation transpires slower, leading to delayed phase separation (liquid–liquid demixing) [7, 15].
MD membranes are typically fabricated from hydrophobic polymers like PP (polypropylene), PTFE (polytetrafluoroethylene), and Polyvinylidene fluoride (PVDF) [16]. Among these materials, PVDF, a semi-crystalline polymer renowned for its exceptional physical and chemical attributes and robust thermal stability, has garnered significant interest. PVDF membranes are commonly produced through NIPS [17]. Various factors in the NIPS technique influence the structure of the membrane, including the composition of the dope, additives, coagulation medium, bath temperature, and solvent strength. This approach typically yields asymmetric membranes with a finger-like morphology in the skin layers near the interface with the nonsolvent [9, 18]. Numerous investigations were conducted to identify suitable nonsolvents. Multiple investigators have proven that the utilization of weaker nonsolvents changes the direction of the phase separation process, resulting in a delay in liquid–liquid demixing and facilitating polymer gelation/crystallization to occur before reaching the binodal curve [19].
PVDF is a semicrystalline piezoelectric polymer with crystalline structures that mostly exist in at least three phases: α, β, and γ. The β phase has an all-trans planar zigzag conformation (TTTT), the α phase has a trans-gauche twist conformation (TGTG′), and the γ phase is an intermediate conformation between the β and α phases [20].
In the current study, a solution consisting of 10% Poly(vinylidene fluoride-co-hexafluoropropylene) (PVDF-HFP) in 85% N, N-Dimethylformamide (DMF) and 5% Tetraethyl orthosilicate (TEOS) was utilized to fabricate an omniphobic membrane exhibiting a contact angle exceeding 120°. The optimal PVDF-HFP, TEOS, and DMF concentrations were selected based on comprehensive rheological investigations outlined in our recently published research [21]. The membrane synthesis was achieved by employing the isothermal immersion-precipitation method. Various characterization techniques were used to evaluate the resulting membrane. The study's primary objective was to develop a highly omniphobic membrane with a porous structure, high surface roughness, enhanced liquid entry pressure (LEP), and maximal contact angle.
Experimental
Materials
PVDF-HFP Solef®21216 from Solvay Specialty Polymers in Bollate, Italy, with respective molecular weights of 600,000 g.mol − 1, underwent a brief drying process at around 60 °C before utilization to eliminate any residual moisture. Procured from Sigma-Aldrich, Tetraethyl orthosilicate 99%, along with the 99.5% pure solvent N,N-Dimethylformamide (DMF) [HCON(CH3)2; weight-average molecular weight 580.14 g/mol] from Sigma–Aldrich (Schnelldorf, Germany) as the solvent, and 1H,1H,2H,2H-Perfluorodecyltrimethoxysilane (PFDTMS) from Sigma-Aldrich (Germany) [C13H13F17O3Si; average molecular weight 610.30 g/mol] were utilized while hexane from Sigma-Aldrich (Germany) with average molecular weight 86.18 g/mol was used as a solvent for PFDTMS.
Preparation of polymer solution
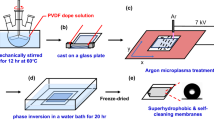
Utilizing Teflon mechanical stirring, PVDF-HFP 10% was dissolved in 85% DMF, as depicted in Fig. 1. Careful drying of the copolymer was conducted to prevent polymer degradation while maintaining dissolution temperatures below 50 °C. The dissolution process lasted between 30 min to 3 h in the absence of TEOS, and this duration was extended when 5% TEOS was introduced, as observed by Li-Yun et al. [22]. Transparent and homogeneous solutions were obtained by following the method [23, 24]. The procedure commenced with the weighing of PVDF-HFP, followed by the addition of DMF and TEOS. Subsequently, the glass beaker was sealed, and PVDF-HFP was dissolved employing a magnetic stirrer. The outcome of these sequential steps was a transparent and uniform solution.

Diagram illustrating the formation of the PVDF-based solution
Membrane fabrication
The membrane was fabricated using the NIPS technique. A hermetically sealed bottle prepared a homogeneous PVDF-HFP, TEOS, and DMF solution. Before casting the membrane, the glass plate and the casting knife were preheated to 80 °C for 10 min to ensure uniform temperature distribution. Subsequently, a thin film of the PVDF-HFP solution, with a thickness of 300 µm, was evenly spread onto the surface of the glass plate. The cast film was promptly submerged in a water bath at 25 °C to facilitate the precipitation of PVDF-HFP-TEOS and promote membrane formation through complete phase separation. The resulting membrane underwent a process of drying within an oven maintained at a temperature of 30 °C for a duration lasting 24 h, aimed at eliminating any residual moisture content and consolidating its structural integrity.
Following this initial stage, a subsequent post-heat treatment procedure was carried out to eradicate any remaining solvent traces from the membrane. This was achieved by subjecting them to an oven that operated at 50 °C for one hour. Subsequently, the membrane, positioned between two glass components, underwent a heating process within the confines of an oven set at a temperature of 110 °C, spanning the entirety of a full day. A secondary round of heat exposure was implemented to continue the treatment process, involving the temperature elevation to 145 °C while applying pressure for one hour. Upon the release of pressure, a gradual reduction in temperature ensued, decreasing by increments of 10 °C per hour until reaching a stable temperature of 100 °C. The membrane was then allowed to remain within the oven environment for an additional day to facilitate further processing. After this stage, a solution consisting of 1% v/v PFDTMS was prepared, following which a measured quantity of 5 mL of this solution was administered onto the surface of the membrane. The membrane treated with PFDTMS was subsequently positioned within an oven operating at a temperature of 60°, with the intent of allowing a timeframe of three hours for the evaporation of hexane.
Characterization of membrane
Multiple techniques and mathematical relationships were used to characterize the membrane's chemical and physical characteristics.
Measurement of mean thickness, mean pore size, liquid entry pressure, porosity, and contact angle of the membrane
The mean thickness of the membrane was determined utilizing a Mitutoyo new model 293–230-30 Digimatic Micrometer. Measurements were obtained from various points on the membrane, and the resulting mean values were computed. Utilizing ImageJ software, the mean pore size of the membrane was evaluated. To assess the LEP of all membranes, a cylindrical pressure filtration cell with a surface area of 25.12 cm2 was utilized. The membrane was situated on the lower surface of the filtration cell and compressed with air and 100 mL of distilled water as the feed. The pressure was incrementally raised from 0.1 bar every 5 min or 30 s based on water flux until it reached 2.5 bar. Subsequently, the pressure was decreased by 1 bar at the same intervals until returning to the initial pressure. Before pressure elevation, the volume of water that traversed the membrane was precisely quantified in both forward and reverse directions. Liquid entry pressure, known as LEP, signifies the pressure at which the initial water droplet permeates the membrane and emerges on the permeate side [25, 26]. By immersing 1 cm2 of each membrane in kerosene at ambient temperature for 24 h, the porosity of the omniphobic membrane was initially determined. After eliminating any excess kerosene from the surfaces of the membrane, the wet weight was quantified, and the dry weight was obtained following the desiccation of the membrane segments in an oven at 50 °C for 24 h. Subsequently, the porosity (P%) was evaluated using a specific mathematical formula:
The wet and dry weights of the membrane, expressed in grams as Ww and Wd, respectively, were identified. The density of the solvent (kerosene) employed for immersing the membrane was denoted as ρs (expressed in grams per cubic centimeter). Conversely, the thickness of the polymer (PVDF) was represented as ρp. In the process of calculating the porosity, the wet and dry weights of the membrane were ascertained utilizing kerosene [27]. The membrane surface's hydrophobic and omniphobic characteristics were evaluated by determining the water and water–ethanol contact angle using an optical tensiometer device, Theta. Distilled water and ethanol solutions of varying concentrations were employed for each measurement. Three measurements were conducted for each membrane's contact angle to minimize experimental discrepancies, and subsequently, the average value was presented.
Study of surface chemical composition and functional groups
The membrane surface chemical compositions and functional groups were analyzed using Cary 360 Fourier-transform infrared spectroscopy (FTIR). The FTIR spectra were obtained through the attenuated total reflection (ATR) method, covering the wavelength range of 4000–400 cm−1. X-ray diffraction analyses were conducted on a Bruker D8 Advance instrument employing Cu Kα radiation (wavelength 1.5418 Å) at 40 kV and 40 mA. Data was acquired over 2θ values from 10◦ to 80◦, with a scanning rate of 1.5 degrees/min.
Study of surface morphology and elemental mappings
The top surface of the membrane was examined using scanning electron microscopy (SEM) with a Merlin 21 Zeiss Gemini 2 instrument. The percentage of each element in the membrane was determined using energy-dispersive X-ray spectroscopy (EDS).
Results and discussion
Characterization of the surfaces of the membrane
The LEP of a membrane shows how much pressure is needed for a liquid to start entering its pores. The LEP of the membrane made of PVDF-HFP, TEOS, and PFDTMS is 1.1 bar, as shown in Table 1, which is relatively high and it can be attributed to a combination of factors. Primarily, the increase in hydrophobicity resulting from the presence of PFDTMS plays a crucial role in enhancing the overall performance of the membrane. The specific pore structure and size distribution within the membrane also contribute significantly to its observed LEP value. Furthermore, the silica network within the membrane matrix further enhances its LEP, thus influencing its overall effectiveness. When TEOS is incorporated into the membrane formulation, it initiates the introduction of silica particles. These particles play a pivotal role in enhancing the hydrophobic characteristics of the membrane while simultaneously filling the interstices within the membrane matrix, thereby effectively diminishing the size of pores within the structure. The rationale behind this reduction lies in the fact that the silica nanoparticles, despite elevating the hydrophobic nature of the membrane, also impose physical constraints on the available volume within the membrane, consequently leading to a decrease in the dimensions of the pore apertures. Moreover, the use of PFDTMS coating aims to bolster the hydrophobic attributes of the membrane by adding lengthy perfluorinated chains. This supplementary modification aimed at enhancing hydrophobicity complements the actions of TEOS, resulting in a compounding effect on the constriction of the pores. The fluorinated chains originating from PFDTMS, in conjunction with the silica nanoparticles derived from TEOS, act synergistically to diminish pore size by physically obstructing spaces and fostering the development of a denser membrane structure. Consequently, the combined influence of TEOS and PFDTMS culminates in an elevation of hydrophobicity and a reduction in pore dimensions owing to the coexistence of fluorine and silica functional groups. Despite the potentially larger dimensions of the silica nanoparticles, their role in reducing pore size is facilitated by their occupation of space within the membrane matrix, ultimately resulting in the observed decline in the average pore size. The porosity of the omniphobic membrane was 43.67%. This porosity value may be due to some large and irregular pores that cause low porosity. The increase in contact angle values observed following the addition of TEOS and PFDTMS to the membrane composition can be attributed to the development of a strong silica network within the matrix, enhancement of hydrophobic properties, alteration of surface morphology, reduction in surface free energy, and the synergistic interaction between these two additional agents. These factors are pivotal in enhancing water resistance and heightening contact angle measurements, evident within the membrane's architecture. When contact angle analysis was performed with a solution of 5%,10%, 15%, and 20% ethanol, the contact angle of the membrane reduced to 118°,107°,99°, and 92°, indicating that the membrane exhibited significant resistance and had an omniphobic character, as the contact angle did not drop below 90°. However, the membrane became wet when the ethanol concentration was increased beyond 20%. The observed reduction in the contact angle of the omniphobic membrane from 130° to 92° following treatment with a 20% ethanol solution indicates that ethanol significantly influences the surface properties of the membrane. Ethanol, as a polar solvent, can engage with the membrane's surface chemistry, potentially resulting in modifications to the fluorinated layer or affecting the spatial distribution of fluorinated substances like PFDTMS. Such interactions may decrease the membrane's hydrophobicity, which can be attributed to the leaching of the fluorinated elements. Overall, this experiment demonstrates that the membrane possessed omniphobic characteristics.
Study of chemical composition and functional groups
The findings from the FTIR analysis of various nanofibers are illustrated in Fig. 2. The FTIR peaks in the spectra provide valuable insights into the membrane's functional groups and crystalline phases. Evident peaks observed at wavenumbers 409 cm⁻1, 485 cm⁻1, and 532 cm⁻1 are linked to bending modes of Si–O-Si, signifying the existence of silicate frameworks derived from TEOS. Furthermore, peaks identified at 613 cm⁻1 and 871 cm⁻1 are associated with CF2 bending and stretching vibrations originating from PVDF-HFP. The spectral features at 760 cm⁻1 and 794 cm⁻1 signify CH2 rocking and CF2 bending vibrations within PVDF, with supplementary contributions from Si–C vibrations in PFDTMS. The discernible peak at 974 cm⁻1 is attributed to Si–O stretching from TEOS or PFDTMS. Moreover, the peaks registered at 1066 cm⁻1 and 1073 cm⁻1 represent Si–O-Si asymmetric stretching, thereby validating the presence of silicate networks and CF3 stretching derived from PVDF-HFP or PFDTMS. The peaks at 1173 cm⁻1 and 1175 cm⁻1 also indicate CF3 stretching vibrations originating from PVDF-HFP or PFDTMS, potentially involving PVDF-HFP. The distinctive peak recorded at 1398 cm⁻1 is likely associated with CF2 and CH2 bending vibrations emanating from PVDF-HFP. About the crystalline structures of PVDF, the peaks at 613 cm⁻1, 760 cm⁻1, and 794 cm⁻1 suggest the presence of the α phase of PVDF, as these peaks are characteristic of CF2 bending and CH2 rocking vibrations specific to this phase, the peak at 840 cm⁻1 shows the existence of the β phase. Notably, definitive peaks of PVDF are absent, specifically indicative of the γ phase (e.g., around 812 cm⁻1, 833 cm⁻1, 1234 cm⁻1).

The FTIR spectra of the omniphobic membrane
The XRD spectra of the PVDF-HFP membrane exhibited a high level of conformity with the findings from the FTIR analysis. In this regard, the XRD spectra unveiled distinct peaks corresponding to the crystalline nonpolar α-phase and the amorphous polar β-phase, aligning well with the revelations from the FTIR spectra as indicated in previous studies. A detailed depiction of the XRD spectra of the membrane can be observed in Fig. 3. Noteworthy peaks at 17.6 and 18.4 were identified, which can be attributed to the nonpolar α-phase. This significant discovery illustrates the predominant existence of the α-phase within the PVDF-HFP composition. It is essential to note that the presence of the α-phase does not imply the exclusive nature of this phase within the PVDF-HFP structure, as evidenced by the emergence of the β-phase peak at 20.9. Consequently, according to previous research findings, the PVDF-HFP membrane exhibited distinct peaks associated with both the α-phase and the amorphous polar β-phase [27,28,29,30]. Ultimately, the FTIR analysis effectively validated the outcomes of the XRD analysis.

XRD pattern of PVDF-HFP-TEOS-PFDTMS
Morphological studies of membrane
The SEM image prominently shows in Fig. 4 a significant increase in the roughness of the observed membrane. This observed increase in roughness can be related to the impact of two distinct contributing factors. Primarily, the addition of TEOS into the membrane matrix leads to the dispersion of silica particles (Si–O-Si), and these particles might have acted as active sites for the fluorinated layer of PFDTMS to be attached on PVDF-HFP thereby causing an overall elevation in roughness. Additionally, the coating of PFDTMS introduces more silica particles into the membrane, further amplifying the observed roughness. Furthermore, the findings obtained from analyzing the contact angle, LEP, and pore size collectively suggest a significant relationship between the presence of silica and the increase in membrane roughness. Particularly, silica particles from both TEOS and PFDTMS with PVDF-HFP contribute to the rise in roughness and decrease in pore size, ultimately resulting in an increased LEP. Consequently, there is a noticeable escalation in the overall roughness of the membrane, accompanied by a corresponding increase in the contact angle due to the combined influence of these factors. Abid et al. worked on fabricating an electrospun membrane with the same composition and observed increased roughness [30]. Moreover, the results obtained from the EDS analysis exhibit a prominent peak in Fig. 4 at the energy level of 1.7 corresponding to the Kα, indicating silicon's existence. These findings validate silica's presence within the membrane material's structural composition.

Scanning electron microscopy images of the surfaces of PVDF-HFP-TEOS-PFDTMS (a) and EDX spectra of PVDF-HFP-TEOS-PFDTMS
Conclusion
In the present investigation, the NIPS technique successfully produced an omniphobic membrane using PVDF-HFP, TEOS, and PFDTMS. The membrane exhibited a notably elevated contact angle of 130°, and the contact angle of the membrane reduced from 130° to 92° with 20% ethanol solution, indicating that the membrane exhibited significant resistance and had an omniphobic character. The reduction in contact angle with ethanol also suggests that ethanol may have caused the fluorinated layer of PFDTMS to leach out. Furthermore, the LEP was quantified at 1.1 bar, underscoring its appropriateness for intricate separation procedures requiring high performance. However, due to some large and variable pores in the membrane, the LEP was not achieved above 2 bar. Analysis of the membrane revealed a porosity of 43.2%, accompanied by a mean pore diameter of 84.19 nm. Through XRD assessment, it was ascertained that the membrane structure encompassed alpha and beta phases. These outcomes imply that the developed membrane possesses highly advantageous characteristics suitable for executing membrane distillation processes, thereby presenting a viable and promising remedy for addressing the challenges encountered in water purification.
Data Availability
The datasets generated and/or analyzed during the current study are available from the corresponding author upon reasonable request.
References
Abid MB et al (2023) Desalination technologies, membrane distillation, and electrospinning, an overview. Heliyon 9(2):e12810
Abid MB, Wahab RA, Gzara L (2023) Membrane Distillation for Desalination and Current Advances in MD Membranes. J Appl Membr Sci Technol 27(2):39–88
Pagliero M et al (2020) Novel hydrophobic PVDF membranes prepared by nonsolvent induced phase separation for membrane distillation. J Membr Sci 596:117575
González D, Amigo J, Suárez F (2017) Membrane distillation: Perspectives for sustainable and improved desalination. Renew Sustain Energy Rev 80:238–259
Guillén-Burrieza E et al (2015) Techno-economic assessment of a pilot-scale plant for solar desalination based on existing plate and frame MD technology. Desalination 374:70–80
Zuo D-Y et al (2006) Influence of alcohol-based nonsolvents on the formation and morphology of PVDF membranes in phase inversion process. Chin J Polym Sci 24(03):281–289
Tao M-M et al (2013) Effect of solvent power on PVDF membrane polymorphism during phase inversion. Desalination 316:137–145
Gopi G, Arthanareeswaran G, Ismail A (2019) Perspective of renewable desalination by using membrane distillation. Chem Eng Res Des 144:520–537
Basko A et al (2023) Mechanism of PVDF membrane formation by NIPS revisited: effect of precipitation bath nature and polymer-solvent affinity. Polymers 15(21):4307
Yu H et al (2023) Chemical cleaning and membrane aging of poly (vinylidene fluoride)(PVDF) membranes fabricated via non-solvent induced phase separation (NIPS) and thermally induced phase separation (TIPS). Sep Purif Technol 313:123488
Tai ZS et al (2023) Critical review on membrane designs for enhanced flux performance in membrane distillation. Desalination 553:116484
Kim A et al (2023) Review on thin-film nanocomposite membranes with various quantum dots for water treatments. J Ind Eng Chem 118:19–32
Buonomenna M et al (2007) Poly (vinylidene fluoride) membranes by phase inversion: the role the casting and coagulation conditions play in their morphology, crystalline structure and properties. Eur Polymer J 43(4):1557–1572
Sukitpaneenit P, Chung T-S (2021) Molecular elucidation of morphology and mechanical properties of PVDF hollow fiber membranes from aspects of phase inversion, crystallization, and rheology. Hollow Fiber Membranes. Elsevier, pp 333–360
Mahmoudi Z (2023) Microencapsulation: Phase inversion precipitation. Principles of Biomaterials Encapsulation, vol One. Elsevier, pp 443–457
Samadi A et al (2023) Engineering antiwetting hydrophobic surfaces for membrane distillation: A review. Desalination 563:116722
Panicker SS, Rajeev SP, Thomas V (2023) Impact of PVDF and its copolymer-based nanocomposites for flexible and wearable energy harvesters. Nano-Struct Nano-Objects 34:100949
Tavajohi N, Khayet M (2024) Polymeric Membrane Formation by Phase Inversion. Elsevier
Young T-H et al (1999) Mechanisms of PVDF membrane formation by immersion-precipitation in soft (1-octanol) and harsh (water) nonsolvents. Polymer 40(19):5315–5323
Shaulsky E et al (2017) Post-fabrication modification of electrospun nanofiber mats with polymer coating for membrane distillation applications. J Membr Sci 530:158–165
Abid MB et al (2024) Effect of TEOS on the rheological properties of polyvinylidene fluoride and poly (vinylidene fluoride-co-hexafluoropropylene). Polimery 69(1):44–51
Nthunya LN et al (2019) A review of nanoparticle-enhanced membrane distillation membranes: membrane synthesis and applications in water treatment. J Chem Technol Biotechnol 94(9):2757–2771
Ribeiro C et al (2018) Electroactive poly (vinylidene fluoride)-based structures for advanced applications. Nat Protoc 13(4):681–704
Abid MB et al (2024) Effect of TEOS on the rheological properties of polyvinylidene fluoride and poly(vinylidene fluoride-co-hexafluoropropylene). Polimery 69(1):44–51
Huo P, Zhong C-T, Xiong X-P (2021) Tailoring morphology of PVDF-HFP membrane via one-step reactive vapor induced phase separation for efficient oil-water separation. Chin J Polym Sci 39:610–619
de Sousa Silva R et al (2021) Membrane distillation: experimental evaluation of liquid entry pressure in commercial membranes with textile dye solutions. J Water Proc Eng 44:102339
Albiladi A et al (2023) Electrospun poly (vinylidene fluoride-co-hexafluoropropylene) nanofiber membranes for brine treatment via membrane distillation. Polymers 15(12):2706
Polat K (2020) Energy harvesting from a thin polymeric film based on PVDF-HFP and PMMA blend. Appl Phys A 126:1–8
Abbas D et al (2022) Active solution heating and cooling in electrospinning enabling spinnability from various solvents. J Appl Polym Sci 139(31):e52730
Abid MB et al (2024) Surface modification for enhanced Anti-Wetting properties of electrospun omniphobic membranes in membrane distillation. J Appl Polym Sci 141(31):e55739
Acknowledgements
The authors extend their appreciation to the Deputyship for Research and Innovation, Ministry of Education in Saudi Arabia for funding this research work through the project number (751).
Funding
The author declares no sources of funding.
Author information
Authors and Affiliations
Contributions
Monis Bin Abid: The individual led data curation, formal analysis, funding acquisition, investigation, methodology, resources, and original draft writing. Lassaad Gzara: This individual provided supporting roles in data curation and methodology. Roswanira Abdul Wahab: Allocation of resources and visualization were equally shared responsibilities for this individual. Iqbal Ahmed Moujdin: The individual took the lead in funding acquisition and methodology while sharing equal responsibility for software. Nadeem and Faridah: These individuals led the investigation process. Mohamed Abdel Salam: This individual was involved in data curation.
Corresponding author
Ethics declarations
Ethical approval
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Abid, M.B., Wahab, R.A., Gzara, L. et al. Advanced omniphobic membrane: Fabrication via NIPS method using PVDF-HFP, TEOS, and PFDTMS. J Polym Res 31, 267 (2024). https://doi.org/10.1007/s10965-024-04121-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-024-04121-1