
Abstract
Polymer dielectrics with synergistic large dielectric constant (ε′) and high breakdown field strength (Eb) have important applications in electronics device and electrical industry. In this work, to enhance the integrated dielectric characteristics of lead zirconate titanate (PZT)/poly(vinylidene fluoride) (PVDF), an insulating magnesia (MgO) shell was constructed on the surface of PZT via a facile chemical precipitation, and the prepared core@shell structured PZT@MgO particles were composited with PVDF to anticipate both high ε′ and Eb but low loss. We explored how the filler loading and MgO shell thickness, frequency affect the dielectric performances of PZT/PVDF composites. The results confirm that the PZT@MgO/PVDF composites show simultaneously improved ε′ and Eb along with low loss over the pristine PZT/PVDF because the MgO interlayer induces multiple-scale polarizations in PZT@MgO/PVDF and clearly boosts the Eb due to markedly prohibited charge injection and migration and electrical branch growth. The optimized ε′ and Eb in composites can be realized by controlling the MgO shell thickness. The theoretic fitting of experimental results by the Havriliak-Negami equation further uncovers the MgO shell’ impact on the polarization mechanism and expounds the inhibiting effect on carrier migration across the composites. The resulting PZT@MgO/PVDF composite dielectrics having both high ε′ and Eb but extremely low loss, display appealing uses in the electrical industries.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Recently, numerous experimental and theoretical efforts focus on dielectric materials with high dielectric permittivity (ε′), low dissipation factor (tanδ) and high breakdown strength (Eb) for civil as well as military power electronics devices, such as medical-defibrillators, power grids, and hybrid electric vehicles (HEVs) [1, 2], etc. In the two common types of dielectrics, traditional dielectric ceramics deliver considerably high ε′ and relatively low loss or tanδ, like barium titanate (BT) and barium strontium titanate (BST), but their low Eb of 200-400 kV/cm and high sintering temperature become a serious bottleneck for high voltage power equipment. While polymer dielectrics have gained tremendous momentum in the study of electrical insulating substances by distinct advantages of outstanding Eb, extra-low tanδ, light weight and ease of processing. However, the rather low ε′ (~ 10) of polymers limits their further extensive applications in energy storage device [3-5]. As a consequence, the current development needs of microelectronic industry cannot be met by a single dielectric material. In order to settle this problem, incorporation of inorganic fillers into polymers to prepare composite dielectrics is one of the preferred methods [6-8].
For the moment, there are two different types of comparatively easy approaches to acquire high ε′ polymer dielectrics. One way is dispersing conductive fillers into insulating polymer matrix, such as carbon-based compounds including carbon black, carbon nanotubes (CNT), graphite, and metal materials (copper, zinc, nickel and so on.) [9-11], etc. The ε′ of polymer composites containing conductive fillers suddenly surges as the filler loading approaches to the percolation threshold (fc), but the remarkably high ε′ is always accompanied with a large loss because of the formation of conductive paths, thereby leading to increased leakage conductivity [3, 5]. Another effective way is doping polymers with giant ε′ ceramic fillers, including BT, BST, SrTiO3, CaCu3Ti4O12, etc. Theoretically speaking, excellent dielectric properties could be realized in the ceramic/polymer composites attributive to the combination of high ε′ of the fillers and large Eb of the polymers. But in reality, the high ε′ induced by high concentration ceramic fillers is always at the expense of deteriorative Eb due to the large contrast in both ε′ and conductivity between ceramic fillers and polymers, as apart from the poor interfacial compatibility and defects [12-14]. For example, Ma et al. integrated the BT into polyvinylidene fluoride (PVDF), confirming that the BT/PVDF has a higher ε′ than PVDF, but the Eb greatly decreases from 150.6 kV/mm of pure PVDF to 58.5 kV/mm of 30 vol% BT/PVDF [15].
Currently, efforts have been made to compensate the decreased Eb, the surface encapsulation by an insulating layer on ceramic or conductive particles is the most widely adopted strategy. Typical insulating shells are divided into organic shell including polydopamine (PDA) [16], polystyrene [17], polymethyl methacrylate [18] and inorganic shell, such as Al2O3 [19], SiO2 [20], SiC [21]. For organic shell, Xie et al. created a novel core-shell structured nanofiller by grafting hyperbranched aromatic polyamide (HBP) onto the surface of BT, and confirmed that the BT-HBP filler contributes to an unexpected Eb for the composites when compared to raw BT [22]. Fan et al. studied the Eb of polylactic acid (PLA) composites containing BT, BT@PDA and BT@PDA@PLA, and found that contrasted to BT/PLA and BT@PDA/PLA, the double-shell BT@PDA@PLA/PLA composites shows significantly enhanced Eb [23]. Although the organic coating with low ε′ is effective in enhancing the interfacial compatibility between the fillers and the matrix, and increasing the Eb in composites, it simultaneously weakens the interfacial polarization (IP) effect to decrease the ε′ to a certain degree [13, 24, 25]. While by introducing a thin inorganic interlayer with moderate ε′ onto the surface of ceramic or conductive fillers can not only construct multiple interfaces in the composites, thus providing more probability for enhanced IP, but also serve as an effective dielectric buffer layer subsequently reducing the diversity in ε′ between fillers and matrix, and blocking the migration of charge carriers, thereby giving rise to remarkably increased Eb.
Inorganic magnesia (MgO) insulation shell has attracted great interest in virtue of its high forbidden bandwidth (~7.8 eV), large resistivity and appropriate ε′ (~10) very close to that of PVDF, as well as extremely low loss (~0.0002). For example, Wang et al. reported that a novel BaTiO3@MgO core-shell nanostructure introduced into poly(vinylidene fluoride-hexafluoropropylene) (P(VDF-HFP)) could maintain a large ε′ and increase the Eb [26]. Chen et al. created core-shell structured BT@MgO particles by a simple chemical precipitation process, and compared with the virginal BT/PVDF, as a buffer barrier, the MgO shell not only weakens the localized electric field distortion at the ceramic/polymer interface resulting in improved ε′ and Eb, but also mitigates the mismatched dielectric characteristics in the BT/P(VDF-HFP) substrate [27].
Among ferroelectric ceramic fillers, lead zirconate titanate (Pb(Zr1-xTix)O3, PZT) is the most promising candidate. It is a solid solution of antiferroelectric lead zirconate (PbZrO3) and ferroelectric lead titanate (PbTiO3) [28]. The composition with Zr/Ti ~ 52/48 exhibits favorable dielectric, ferroelectric and piezoelectric property below the curie temperature [29]. However, if PZT particle is straightforwardly used as filler, the high surface energy will make them form strong agglomeration in a matrix, resulting in a sharp decline in the Eb and capacitive performances of composites. Additionally, the large dielectric difference between PZT and polymers will cause local concentration and distortion of the applied electric field near the ceramic/polymer interface [14]. As a result, until now, it is still a great challenge to obtain polymeric composites with both high ε′ and Eb towards energy storage purposes.
Therefore, this work intends to prepare core-shell structured PZT particles through a chemical precipitation method, and then the resulting PZT@MgO particles were composited with PVDF to purposefully obtain morphology-controllable advantageous dielectric characteristics. And the influences of filler contents and frequency on the ε′, loss, electrical conductivity and Eb of composites are comparatively studied [30]. The findings should provide deep insight into the search for useful core-shell structured PZT@MgO particles and their composites with large ε′ and Eb but small loss.
Experimental
Materials
Lead zirconate titanate (PZT) particles were supplied by Shanghai Dian Yang Industry Co., China. PVDF FR901 was bought from 3F New Materials Co. (Shanghai, China). Magnesium chloride hexahydrate (MgCl2·6H2O, AR grade) were provided by Shanghai Yien Chemical Technology Co., China. Potassium hydroxide (KOH, AR grade) and Sodium lauryl sulfate (CH3(CH2)11SO4Na, AR grade) were obtained from Tianjin Hongyan Chemical Reagent factory, China. N, N-Dimethylformamide (DMF) was purchased from Tianjin Chemical Reagent Co., China. Deionized water was provided from Kemiou Chemical Reagent Co., Ltd., Tianjin, China.
Preparations
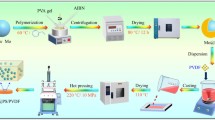
The fabrication method for the core-shell PZT@MgO particles and their PVDF composites is schematically shown in Fig. 1. Firstly, 1 g PZT particles and 1 g CH3(CH2)11SO4Na were added into a three-necked flask that included 200 mL deionized water, and the mixture was ultrasonically treated for 1 h to prepare stable suspension with good dispersion. Then, MgCl2·6H2O with the target amount was added and stirred by magnetic stirring for 30 min. Next, a suitable amount of KOH solution was added and the solution’s pH was changed to fall within the suitable alkalinity range. Subsequently, this mixture was put into a water bath 25 °C with magnetic stirring for 8 h. Then it was washed with absolute ethanol and deionized water repeatedly at least three times, and the samples was put into an oven at 80 °C for 8 h to remove the residual solvent. Finally, the dry sol was crushed with an agate mortar, and calcined in a muffle furnace at 600 °C under air for 2 h to obtain the core-shell structured PZT@MgO particles with diverse MgO loadings.

Schematic showing the preparation of PZT@MgO and their PVDF composites
The PVDF composites with varied fillers were created by hot pressing after a solution mixing process. First, PVDF was entirely dissolved in DMF solvent under agitation at 50 °C for 4 h; and the PZT@MgO particles were dispersed in DMF solvent by an ultrasonic cleaner for 30 min. Next, the dispersed filler and the homogeneous PVDF solution were mixed and stirred for 12 h at room temperature. After that, the majority of the DMF was subsequently removed by casting the mixture onto a clean glass plate and drying it in a vacuum oven at 100 °C for 3 h. Then, it was hot-pressed for 30 min at 220 °C with a pressure of roughly 15 MPa. The obtained samples are labeled as PZT-1 (PZT@1 wt%MgO)/PVDF, PZT-2 (PZT@3 wt%MgO)/PVDF, PZT-3 (PZT@5 wt%MgO)/PVDF, respectively, and the PZT-0 represents the raw PZT/PVDF.
Characterizations
A Fourier transform infrared (FT-IR) spectrometer (Perkin-Elmer, Paragon1000) was used to analyze the surface chemistry of the specimens in the 400-4000 cm−1 wavenumber range. By using a Shimadzu X-ray difractometer-6000 (XRD) equipped with a graphite homochromatic instrument and a Cu anticathode (40 kV, 30 mA, scanning rate 2°/min), the structure of PZT@MgO particles was examined. A JEOL JSM-7000F scanning electron microscopy (SEM) outfitted with an energy dispersive spectroscopy (EDS) equipment was used to examine the appearance of fillers and the broken surface of the composite specimens. The specimens were broken in liquid N2 before being seen under a scanning electron microscope, and a coating of gold was sputter-coated onto the rupture surface.
The samples’ dielectric performance was measured via an Agilent 4294A impedance analyzer in the measuring frequencies from 40 to 107 Hz. The measurement of Eb was implemented at room temperature with 20 mm ball to ball electrode under AC voltage. The specimens were permeated in the silicone oil to prohibit surface discharge and flashovers. The bottom electrode was connected to the ground, and an AC voltage with a ramp of 2 kV/s was received to the upper electrode until the specimens failed.
Results and discussion
Characterizations of PZT@MgO particles
Figure 2a shows the FT-IR spectra for pure PZT and various PZT@MgO particles, respectively. The vibration of -OH groups on the particle surfaces can be connected to the absorption peaks at 3437 cm−1. The PZT@MgO particle presents a new absorption peak at 705 cm−1 than the pure PZT, and the strength of this peak increases when the MgO shell thickness is raised. Meanwhile, compared to the pure PZT, PZT@MgO exhibits much lower absorption peaks at 1096 cm−1 and 2328 cm−1. As a result, the finding shows the victorious coverage of a MgO shell around PZT particle.

a FT-IR spectrum and b XRD pattern of PZT, PZT-1 and PZT-3 particles, c-h EDS and element mapping of the PZT@MgO particles
The XRD patterns for both the pure PZT and the core-shell structured PZT particles are shown in Fig. 2b. Each XRD pattern has three significant peaks at 21.2°, 30.7°, 38.3°, 45.2° and 54.6°, corresponding to the (100), (110), (111), (200) and (211) lattice planes of PZT particle, respectively. Besides, additional diffraction peaks at 42.9° and 62.2° in the PZT@MgO particles arise in comparison to the pure PZT particles and are compatible with the (200) and (220) crystal planes of MgO, respectively. This suggests that a layer of MgO shell is formed on the surface of raw PZT.
As can be shown in Fig. 2c-h, evidently, the results from FT-IR and XRD analyses are similar with the EDS mappings, which show that the surface of samples is composed of Pb, Zr, Ti, Mg, and O elements. Also, the even distribution of the Mg and O elements suggests that a uniform MgO shell has been generated on the surface of raw PZT particles [31]. In contrast to PZT-0/PVDF, the effectively coated MgO shell as an interlayer will have a significant impact on the mechanical, dielectric, and other characteristics of the composites.
Microstructure
Figure 3 shows the TEM images of pristine PZT and PZT@MgO particles. From Fig. 3a, c, on the surface of raw PZT, no noticeable exterior shell can be seen, but as seen in Fig. 3b, d, a shell is clearly visible outside the PZT core, and the shell thickness is around 20 nm, providing direct evidence that the PZT@MgO particle exists. Besides, keeping with the findings of the XRD measurement, the shell exhibits prominent lattice fringes, which is the clearest indication of crystalline MgO.

TEM images of a, c PZT and b, d PZT@MgO particles, respectively
Figure 4 presents the SEM images of pristine PZT and PZT@MgO particles and their PVDF-based composites that include 30 wt% and 50 wt% of fillers, respectively. From Fig. 4a, the diameter of pristine PZT particles is about 2-4 μm, and have a rough surface; but the PZT@MgO particles show a smooth surface because of the presence of a layer of MgO shell, as seen in Fig. 4b. From Fig. 3c-f, the pure PZT particles are dispersed heterogeneously in the PVDF with obvious aggregation because of the weak interfacial interactions between the matrix and filler particles. And yet, the PZT@MgO particles present a more even dispersion in the PVDF, attributive to improved interfacial interactions between the matrix and fillers. The more numbers of hydroxyl groups in PZT@MgO can establish hydrogen bonds with the F atom in the PVDF compared with pure PZT, which enhances the interface compatibility and improves the fillers’ dispersion in the matrix. Thus, the aforementioned findings attest to the successful fabrication of core-shell structured PZT@MgO particles.

SEM images of a PZT-0, b PZT-2, c 30 wt% PZT-0/PVDF, d 30 wt% PZT-2/PVDF, e 50 wt% PZT-0/PVDF and f 50 wt% PZT-2/PVDF composites
Dielectric properties
According to the filler quantity and frequency, Fig. 5 shows the dielectric characteristics of PVDF with varied loadings of PZT and PZT@MgO particles. In short, the ε′ of all composites shows definite decrease with increasing frequency owing to the disappearance of IP in the high frequency range and the hysteresis of dipole polarization in the high frequency range. Meanwhile, there is enough time to orient with the frequency alternation because all specimens’ ε′ exhibits an asthenic frequency dependency and finishes in a particular constant value between 40 and 105 Hz [13, 32]. Compared to pure PVDF, the addition of PZT and PZT@MgO fillers can increase the ε′ of composites, and this phenomenon can be more obviously strengthened by increasing their content because of the Maxwell-Wagner-Sillars (MWS) phenomenon, which develops at heterogeneous phases with varying electric conductivities and is caused by the filler’s greater polarizability than the polymer’s.

ε′ reliance on frequency and filler content for a PZT-0/PVDF, b PZT-1/PVDF, c PZT-2/PVDF, d PZT-3/PVDF, e ε′ reliance on filler content for the PVDF padded with PZT and PZT@MgO at 100 Hz, f ε′ reliance on frequency for the PVDF padded with PZT-0 ~ PZT-3 at 50 wt% filler content
According to Fig. 5a, the following two processes may be used to explain that the rise ε′ of the PZT-0 as the filler concentration is increased. The low level of the ε′ at filler loadings below 20 wt% is related to the difficulties of producing enough charge carriers and micro capacitors due to the large spacing between PZT particles. The ε′ shows a noticeable improvement when the filler loading increases up to 30 wt% due to the improved IP brought on by the ongoing production of micro capacitors. For instance, the ε′ of PVDF composite with 50 wt% PZT-0 at 100 Hz increases to 16.3 from 12.3 of pure PVDF.
From Fig. 5b-d, PZT-1/PVDF ~ PZT-3/PVDF show the same trend as PZT-0/PVDF, but for the composites containing the same filler contents, the PZT-1/PVDF ~ PZT-3/PVDF composites with MgO shell deliver preferable ε′ compared with PZT-0/PVDF. It should be noticed that as an interface layer, the MgO shell productively promotes and enhances the contribution to the IP in PZT@MgO/PVDF composites, that is to say, there are two types of interfaces in the PZT@MgO/PVDF composites, including the interface between PZT and MgO interface, the interface between MgO and PVDF. The higher ε′ is the results of charges accumulation at the interfaces between the three dielectric components, subsequently leading to enhanced IP. It is speculated that the IP between PZT and MgO is stronger than that between PZT and PVDF, more interfaces may bring about a distinctly increase in ε′. Besides these similar behaviors, different MgO content also has a certain influence on ε′ of composites [33, 34].
As indicated in Fig. 5e, the ε′ for PZT@MgO/PVDF initially improves as the MgO shell thickness rises because of the progressively raised interface compatibility and the contribution to IP from the newly established interfaces between PZT and PVDF, thereby assisting in the improvement of IP. However, the ε′ will decrease as the MgO shell thickness further increases because the likelihood of electrons tunneling and hopping across the shell decreases, which suppresses the IP effect. As shown in Fig. 5f, what is notable is that, when compared to PZT-0/PVDF, PZT-1/PVDF, and PZT-3, PZT-2 endow the PVDF with the greatest ε′ at low frequency, showing that the IP may be induced to an ideal level by adjusting the MgO shell thickness for excellent dielectric performances.
Figure 6 displays the tanδ as a function of frequency and filler content for PZT-0 ~ PZT-3 filled composites. Distinctly, from Fig. 6a-d, the tanδ of all specimens reduces in the frequency range from 40 to 104 Hz, and then increases considerably over 105 Hz. The tanδ at low frequencies is mainly caused by the interface polarization friction between fillers and the matrix, as a result of macroscopical heterogeneous interfaces of the different diphase structures. Simultaneously, the obviously high tanδ at high frequencies mainly comes from the dielectric relaxation derived from the C-F dipole directional polarization in PVDF matrix.

tanδ reliance on frequency and filler content for a PZT-0/PVDF, b PZT-1/PVDF, c PZT-2/PVDF, d PZT-3/PVDF, e tanδ reliance on filler content for various composites at 100 Hz, f tanδ dependence on frequency for the PVDF with PZT-0 ~ PZT-3 at 50 wt% filler content
According to Fig. 6a, the PZT concentration at low frequencies essentially has no influence on the fluctuation in tanδ for the PZT-0/PVDF composites. However, it is found that an important improvement of tanδ is seen at high frequency, and the correlation with filler loading is enhanced. This effect may be attributed to the direct contact and agglomeration of highly concentrated PZT in the PVDF, which results in the buildup of charge carriers and the electron conduction process. From Fig. 6b-d, it can be found that the tanδ of the PZT-1/PVDF ~ PZT-3/PVDF still retains a lower value. This mirrors that the MgO interlayer has the ability of lowering tanδ of the composites. The causes are often attributed to the MgO insulating layer placed on the surface of PZT cores, which serves as an interlayer and prevents direct contact between adjacent PZT powders, thereby productively restraining the mobility of free charge carriers. To further analyze the tanδ of the composites, the tanδ of PZT-0/PVDF ~ PZT-3/PVDF are also summarized in Fig. 6e-f, compared with PZT-0/PVDF, the tanδ color of PZT-1/PVDF ~ PZT-3/PVDF display almost the same color as the pristine PZT-0/PVDF composites, i.e., the tanδ only slightly increases.
For the PZT/PVDF and PZT@MgO/PVDF composites, Fig. 7 shows how the conductivity of alternate current (AC) varies with frequency and filler amounts. As seen in Fig. 7a-d, the AC conductivity of the composites rises stably with the increase of frequency and displays almost an independent of the filler concentration. Compared with PZT-0/PVDF, the electrical conductivity of PZT-1/PVDF ~ PZT-3/PVDF decreases slightly, which suggests that the MgO insulating shell could serve as a barrier interlayer between the raw PZT particles to prevent them from contacting with each other subsequently leading to restrained leakage current in the composites [35-37]. As indicated in Fig. 7e, the conductivity of the composites decreases significantly in PZT-3/PVDF under the same filler content. It’s suggested that PZT-2 filler exhibits the strongest inhibitory influence on the electronic conduction processes and the movement of free charge carriers of the composites. In Fig. 7f, the MgO interlayer can prevent long-range charge carrier transfer, restrict mobility around the PZT particle surface, and effectively prevent the formation of current channel, thereby reducing the AC conductivity of composites.

Electric conductivity dependence on frequency and filler content for a PZT-0/PVDF, b PZT-1/PVDF, c PZT-2/PVDF, d PZT-3/PVDF, e Electric conductivity of raw PVDF or various PZT@MgO/PVDF at 100 Hz, f Schematic illustration of the charge distribution and propagation pathway in PZT/PVDF and PZT@MgO/PVDF composites, respectively
To learn more about the polarization process in the PVDF with PZT@MgO with various shell thicknesses, single polarization must be separated from multiple polarizations. The Havriliak-Negami (H-N) equation is used to simultaneously fit the imaginary portion of dielectric constant (ε′′) vs. real component of dielectric constant (ε′) as shown below [38]:
where \(\Delta {\varepsilon }_{i}\) is the relaxation strength values, and the \({\tau }_{HNi}\) is the characteristic relaxation time, \({\sigma }_{dc}\) and \({\varepsilon }_{0}\) are the conductivity loss and ε′ in vacuum (8.85 × 10–12 F·m−1), respectively. The parameters \({\alpha }_{i}\) and \({\beta }_{i} [0<{\alpha }_{i}; {\alpha }_{i}{\beta }_{i}<1]\) define the symmetrical and asymmetrical broadening of the distribution of relaxation times, respectively, and the i stands for the various relaxations involved in dielectric spectra.
Figure 8 depicts the deconvolution of the PVDF with PZT-0 ~ PZT-3 at 50 wt%. Simultaneously, the dedication of each relaxation mode is drawn. In general, there are two relaxations underlying the dielectric response of the composites. One of these is the segment relaxations from an amorphous PVDF matrix, which is triggered at a frequency of 105-107 Hz, and is consistent with ε′ in Fig. 8a-d. Another is IP occurring between the functional fillers and the matrix.

a-d Dielectric spectra of PZT-0/PVDF ~ PZT-3/PVDF at 50 wt% deconvolutions into distinct relaxations using the H-N equation, e-g summary and evolution of best-fitting parameters with varying shell thicknesses
Large filler polarization results from the charge’s uneven distribution acting as a permanent electric dipole. It is clear from Fig. 8a-d that the ε′′ rises sharply in the low-frequency region, which is thought to represent the long-distance movement of residual charges. It is generally established that IP has an intrinsic relationship with both charge migration and electron displacement. In contrast to charge migration, which is an accurate description of electron transfer between the filler particle and the outer shell (rapid polarization), electron displacement explains the charge diffusion across consecutive filler contacts quite well. It is clear that these two IP effects are undoubtedly the principal determinants of the dielectric characteristics of samples.
Figure 8e-g gives the best-fitting results. The filler polarization is clearly related to the low-frequency relaxation peak intensity (Δε1), which differs from PVDF matrix relaxation behavior, showing a non-linear association with the increase in MgO shell thickness in PZT-0/PVDF ~ PZT-3/PVDF composites in Fig. 8e. The relaxation peak strength and relaxation time prove a tendency to increase and then decrease with raising the shell thickness of MgO in the PZT-0/PVDF ~ PZT-3/PVDF composites. Clearly, the transition from PZT-0/PVDF to PZT-3/PVDF composites exhibits a propensity to first enhance and subsequently decrease the low-frequency relaxation (relaxation-1), which is also indicated in Fig. 6 [39]. The relaxation peak intensity (Δε2) is 14 at high-frequency and the relaxation duration (τ2) is around 10–8 s, which shows a slight variation with the different shell thicknesses of MgO in PZT-0/PVDF ~ PZT-3/PVDF composites in Fig. 8f. It is clear that the PVDF matrix, which is present in the same concentrations in the composites under discussion, is the source of the aforementioned relaxation. Besides, in composites with PZT-0/PVDF ~ PZT-3/PVDF, the shape parameters (α) of all relaxations show a little variation with varying MgO shell thicknesses, pointing to a semblable polarization process in Fig. 8g [40]. In addition, the electric conductivity in PZT-0/PVDF ~ PZT-3/PVDF composites has a trend to decline with increasing the MgO shell thickness, which is consistent with the long-term electron migration being successfully halted by the MgO shell, as seen in Fig. 8g.
The Cole-Cole figures of ε′′ vs. ε′ for PVDF with pure PZT and various PZT@MgO particles are shown in Fig. 9. The Cole-Cole graphs of all composites clearly show two semblable semicircular arcs, which are similar in characteristics. The first little semicircular arc denotes the rapid relaxation associated with the PVDF matrix’s C-F dipole polarization behavior, which takes place in the high frequency range. Additionally, the second great semicircular arc, which appears in the low frequency area, is due to the IP effect. The big semicircle’s degree of deviance from the ε′-axis also serves as a proxy for the magnitude of conductivity loss, as seen in Fig. 9a-d, which similarly mirrors that the loss from conductivity rises with filler concentration. The Cole-Cole equation, which consists of the imaginary and real parts of ε∗, may be written as follows:
where \({\varepsilon }_{\infty }\) stands for the optical ε′ of \({\varepsilon }^\prime\left(\omega \to \infty \right)\), \({\varepsilon }_{s}\) is the electrostatic \({\varepsilon }^\prime\left(\omega \to 0\right)\), \(\omega =2\pi f\) is thought of the angular frequency, τ represents relaxation time, and \(\sigma\) is the electric conductivity [41, 42].

Cole-Cole curves of ε′′ vs. ε′ for PZT-0/PVDF ~ PZT-3/PVDF composites (a-e) containing different filler concentrations
In addition, \(i\frac{\sigma }{\omega }\) in Eq. (2) illustrates the effect of electric conductivity, which has a substantial impact on the geometry of Cole-Cole splines’ big semicircles. In Fig. 9a-d, the electric conductivity raises as the filler content rises. The DC conductivity has a considerable impact on the shape of the curve, which diverges from a semicircle and exhibits a clearly deformed arc-shaped pattern. The Cole-Cole curves' semicircle width does not considerably increase when filler content goes from 10 wt% to 50 wt%, from PZT-0/PVDF composites to PZT-1/PVDF ~ PZT-3/PVDF composites, associating with the growing trend in electric conductivity. In comparison to PZT-0/PVDF, PZT-1/PVDF ~ PZT-3/PVDF composites have a constant improvement in the shell thickness, which significantly increases electrical resistivity as shown in Fig. 5a and suppresses the ε′′ relaxation behavior, resulting in a narrower semicircle and relaxation curve in low frequencies.
The Eb is largest electric field strength that a dielectric material can withstand without losing its insulating properties. Particularly, for dielectric applications, the key factor is to enhance Eb of a composite. As shown in Fig. 10b-e experimentally observed Eb is analyzed with the Weibull statistic:
where P stands for the cumulative probability of electrical failure, and Eb is the characteristic Eb of each sample, expressed as the electric field accumulated to 63.2% under the failure probability, β is the Weibull modulus, quantifying the discreteness of the experimental results [11, 41, 43, 44].

a The Eb changes with different filler loading for composites, b-e the Weibull distribution for various composites, f the schematic representation of the energy gap of PZT@MgO
As shown in Fig. 10a, the Eb of composites tends to reduce with increasing filler concentration, especially at high filler contents. This is because the interfaces between the polymer matrix and the filler may have more structural defects and higher electric field concentrations or distortion. However, the surprise is that the PZT@MgO/PVDF still reveals a higher Eb than that of original PZT/PVDF. Compared to the raw PZT/PVDF, the Eb of the PZT@MgO/PVDF composites has been improved due to the existence of the insulating MgO interlayer [45]. On the one hand, introducing an inorganic MgO shell with an intermediate relative ε′ between the PZT particles and the PVDF reduces and suppresses the local electric field concentration and distortion. The interface can change the distribution and motion of space charges, which confines the mobility of free electrons and excessive current percolation effects, thereby resulting in a reduction in leakage current and tanδ, improving the Eb. On the other hand, as indicated in Fig. 10f, for core@shell structured PZT@MgO, the insulating MgO possesses a wide band gap between the conduction band (CB) and the valence band (VB) about 7.8 eV, and the electron can difficultly jump to CB and establish the conductive network.
Besides, the original PVDF has an Eb of 9.73 kV/mm. With the addition of PZT@MgO, the Eb of the PZT@MgO/PVDF composites first increases to 10.31 kV/mm for PZT-1 and 10.79 kV/mm for PZT-2. However, when PZT@MgO fraction further increases from 3.0 wt% to 5.0 wt%, introducing more structural defects such as interfacial voids in SEM images (Fig. 6), Eb shows a gradual decrease from 10.79 kV/mm to 10.63 kV/mm. The results show that the introduction of high-insulating MgO plays a significant role in the Eb reinforcement of the composites, primarily because of the alleviation of dielectric mismatch and local electric field distortion, and the limitation of electron migration between PZT fillers and PVDF.
In order to further understand the effect of MgO shell on the dielectric properties of the composites, Fig. 11a shows a schematic diagram of the charge and dipole motion at the interface regions for the PZT/PVDF and PZT@MgO/PVDF under an external electric field. As shown in Fig. 11a, the agglomeration phenomenon of PZT in PVDF results in fewer carrier channels in composite. However, for PZT@MgO/PVDF, the MgO interlayer not only improves the dispersion of PZT powder in the polymer, but also enhances the interface polarization degree between the filler and the matrix, clearly increasing both the ε′ and Eb of the composite. Moreover, the MgO shell provides longer carrier channels, which prohibits charge movement in the PZT@MgO/PVDF.

a Schematic representation of charge and dipole movements at the interface area of PZT/PVDF and PZT@MgO/PVDF under an external electric field. Conduction process flowchart in the b PZT/PVDF and c PZT@MgO/PVDF (LUMO is the smallest vacant molecular orbital and HOMO is the largest occupied molecular orbital, EF represents Fermi level)
To learn more about how the MgO shell affects the composite’s Eb, Fig. 11b, c presents a schematic diagram of the conduction process in both PZT/PVDF and PZT@MgO/PVDF composites. In general, both composites have traps corresponding to relaxation 1 and relaxation 2, the interfaces in PVDF’s crystalline/amorphous phases are the places where the relaxation 1of trap 1 forms, and the trap of relaxation 2 may be derived from IP between particles and the amorphous phase of PVDF, where trap 2 is provided by PZT and MgO provides trap 3. For PZT/PVDF (Fig. 11b), the free electric charge carriers in the PZT may be trapped in a trap during migration, and this limits the mobility of the carriers in the PZT/PVDF composites and lowers electrical conductivity since the trapped carriers are unable to continue to take part in the conduction process. It is worth noting that in contrast to raw PZT/PVDF, the trap levels corresponding to relaxations 1 and 2 are both strengthened by the PZT@MgO filler (Fig. 11c). Moveable charges are limited at the interfaces between the PZT-MgO and MgO-PVDF due to the large trapping sites for charges along the extended interfaces that may be provided by the core-shell structured PZT@MgO [46, 47], thus suppressing the conductivity of the composites. Moreover, the MgO with a wide bandgap (~7.8) exhibits excellent electrically insulating properties, reduces the dielectric mismatch and mitigates the local electric field distortion, and inhibits electron migration between PZT and PVDF, thereby enhancing the Eb of PZT@MgO/PVDF composites.
Conclusion
Core@shell structured PZT@MgO particles were fabricated as fillers to fill PVDF by a straightforward chemical precipitation procedure followed by solution mix in order to effectively boost the ε′ and Eb of pure PZT/PVDF. Measurements from the XRD, FT-IR, SEM, and TEM confirm that the PZT’s surface is covered with a layer of MgO shell. Performance tests reveal that in contrast to the pure PZT/PVDF, the PZT@MgO/PVDF composites possess higher ε′ and larger Eb. That is because the use of MgO as a buffer barrier effectively alleviates the dielectric mismatch between PZT particles and PVDF, enhances interfacial compatibility, and promotes the uniform dispersion of fillers in PVDF. In addition, the MgO shell with a large band gap shows excellent insulation properties, inhibits electron migration between PZT and PVDF, thereby improving the Eb of the composites. With tailoring the MgO shell thickness, the composites’ optimum dielectric characteristics can be attained. The theoretic fitting results further uncover the MgO shell’ impact on the polarization mechanism and the migration of carrier charges in the composites. This work manifests that the encapsulation of an MgO shell on the surface of PZT is a favorable way to manufacture the flexible PVDF dielectric composites with high ε′ and Eb, as well as low loss.
References
Zhang N, Liu XR, Zhao SS, Yang ZW (2022) Crystal structures of three transition metal complexes with salicylaldehyde-4-hydroxy phenylacetyl acylhydrazone and their interactions with CT-DNA and BSA. Polyhedron 211
Zhang X, Shen Y, Xu B, Zhang Q, Gu L, Jiang J, Ma J, Lin Y, Nan CW (2016) Giant Energy Density and Improved Discharge Efficiency of Solution-Processed Polymer Nanocomposites for Dielectric Energy Storage. Adv Mater 28(10):2055–2061
Chang LL, Yang J, Liu XR, Yang ZW (2022) Zhao SS (2022) Synthesis, crystal structures and CT-DNA/BSA binding properties of Co(III) and Cu(II) complexes with bipyridine Schiff base ligand. Inorg Chim Acta 532
Zhao D, Zhang Z, Ren J, Xu Y, Xu X, Zhou J, Gao F, Tang H, Liu S, Wang Z, Wang D, Wu Y, Liu X, Zhang Y (2023) Fe2VO4 nanoparticles on rGO as anode material for high-rate and durable lithium and sodium ion batteries. Chem Eng J 451
Huang DS, Liu XR, Zhao SS, Yang ZW (2022) Crystal structures of three transition metal complexes with salicylaldehyde-4-hydroxy phenylacetyl acylhydrazone and their interactions with CT-DNA and BSA. Polyhedron 211
Khanchaitit P, Han K, Gadinski MR, Li Q, Wang Q (2013) Ferroelectric polymer networks with high energy density and improved discharged efficiency for dielectric energy storage. Nat Commun 4(1):2845
Yang Z, Xu TT, Zhang X, Li H, Jia XD, Zhao SS, Yang ZW, Liu XR (2022) Nitrogen–doped carbon quantum dots as fluorescent nanosensor for selective determination and cellular imaging of ClO- Spectrochim Acta Part A Mol Biomol Spectrosc 271
Chen Q, Shen Y, Zhang SH, Zhang QM (2015) Polymer-based dielectrics with high energy storage density. Annu Rev Mater Res 45:433–458
Luo H, Ma C, Zhou XF, Chen S, Zhang D (2017) Interfacial design in dielectric nanocomposite using liquid crystalline polymer. Macromolecules 50(13):5132–5137
Xu TT, Li H, Yang HN, Yang Z, Jia XD, Zhao SS, Yang ZW, Liu XR (2022) Nitrogen-Doped and Surface Functionalized CDs: Fluorescent Probe for Cellular Imaging and Environmental Sensing of ClO-. J Fluoresc 32:1591–1600
Zhao LH, Liao CJ, Liu Y, Huang XL, Ning WJ, Wang Z, Jia LC, Ren JW (2022) A combination of aramid nanofiber and silver nanoparticle decorated boron nitride for the preparation of a composite film with superior thermally conductive performance. Compos Interfaces 29(4):447–463
Yang Z, Li H, Xu TT, Liu XR, Zhao SS, Yang ZW (2020) Azaaromatic Functionalized Rhodamine Based Fluorescent Probes for Selective Dual Channel Detection of ClO1 and Cu2+ in Water Samples and Living Cells. Chem Lett 49(11):1278–1281
Zheng MS, Zhang C, Yang Y, Xing ZL, Chen X, Zhong SL, Dang ZM (2020) Improved dielectric properties of PVDF nanocomposites with core-shell structured BaTiO3@polyurethane nanoparticles. IET Nanodielectrics 3(3):94–98
Zhong SL, Dang ZM, Zhou WY, Cai HW (2018) Past and future on nanodielectrics. IET Nanodielectrics 1(1):41–47
Ma JC, Azhar U, Zong CY, Zhang YB, Xu AH, Zhai CC, Zhang LQ, Zhang SX (2019) Core-shell structured PVDF@BT nanoparticles for dielectric materials: A novel composite to prove the dependence of dielectric properties on ferroelectric shell. Mater Des 164
Zhou WY, Kou YJ, Yuan MX, Li B, Cai HW, Li Z, Chen FX, Liu XR, Heng GH, Chen QG, Dang ZM (2019) Polymer composites filled with core@double-shell structured fillers: effects of multiple shells on dielectric and thermal properties. Compos Sci Technol 181
Zhou WY, Li T, Yuan MX, Li B, Zhong SL, Li Z, Liu XR, Zhou JJ, Wang Y, Cai HW, Dang ZM (2021) Decoupling of inter-particle polarization and intra-particle polarization in core-shell structured nanocomposites towards improved dielectric performance. Energy Stor Mater 42:1–11
Zhou WY, Cao GZ, Yuan MX, Zhong SL, Wang YD, Liu XR, Cao D, Peng WW, Liu J, Wang GH, Dang ZM, Li B (2022) Core-Shell Engineering of Conductive Fillers toward Enhanced Dielectric Properties: A Universal Polariz ation Mechanism in Polymer Conductor Composites. Adv Mater 2207829
Qu MN, Liu QH, Shi F, Lv YQ, Liu H, Mu LH, Liu XF, He JM (2023) Flexible conductive Ag-CNTs sponge with corrosion resistance for wet condition sensing and human motion detection. Colloids Surf, A 656
Pradhan SK, Kumar A, Kour P, Pandey R, Kumar P, Kar M, Sinha AN (2020) Piezoelectric and mechanical properties of PVDF-PZT composite. Ferroelectrics 558(1):59–66
Wang P, Zhang XM, Duan W, Teng W, Liu YB, Xie Q (2021) Superhydrophobic flexible supercapacitors formed by integrating hydrogel with functional carbon nanomaterials. Chin J Chem 39(5):1153–1158
Xie LY, Huang XY, Huang YH, Yang K, Jiang PK (2013) Core-shell structured hyperbranched aromatic polyamide/BaTiO3 hybrid filler for poly(vinylidene fluoride-trifluoroethylene-chlorofluoroethylene) nanocomposites with the dielectric constant comparable to that of percolative composites. ACS Appl Mater Interfaces 5(5):1747–1756
Fan YY, Huang XY, Wang GY, Jiang PK (2015) Core-shell structured biopolymer@BaTiO3 nanoparticles for biopolymer nanocomposites with significantly enhanced dielectric properties and energy storage capability. J Phys Chem 119(49):27330–27339
Zha JW, Yao SC, Qiu Y, Zheng MS, Dang ZM (2019) Enhanced dielectric properties and energy storage of the sandwich-structured poly(vinylidene fluoride-co-hexafluoropropylene) composite films with functional BaTiO3@Al2O3 nanofibres. IET Nanodielectrics 2(3):103–108
Qu MN, Lv YQ, Ge JW, Zhang B, Wu YX, Shen L, Liu QH, Yan M, He JM (2023) Hydrophobic and multifunctional strain, pressure and temperature sensor based on TPU/SiO2-ILs ionogel for human motion monitoring, liquid drop monitoring, underwater applications. Colloids Surf, A 664
Wang P, Zhou D, Guo H, Liu W, Su J, Fu MS, Singh C, Trukhanov S, Trukhanov A (2020) Ultrahigh enhancement rate of energy density of flexible polymer nanocomposites by Core-Shell BaTiO3@MgO Structures as Fillers. J Mater Chem A 8(22):11124–11132
Chen J, Huang FX, Zhang CY, Meng FC, Cao LL, Lin HX (2022) Enhanced energy storage density in poly (vinylidene fluoride-hexafluoropropylene) nanocomposites by filling with core-shell structured BaTiO3@MgO nanoparticals. J Energy Storage 53
Qu MN, Yang X, Peng L, Liu LL, Yang C, Zhao Z, Liu XR, Zhang TJ, He JM (2021) High Reliable Electromagnetic Interference Shielding Carbon Cloth with Superamphiphobicity and Environmental Suitability. Carbon 174:110–122
Newnham RE (1989) Electroceramics. Rep Prog Phys 52(2):123
Chen X, Liang F, Lu W, Zhao Y, Fan G, Wang X (2018) Improved dielectric properties of Ag@TiO2/PVDF nanocomposites induced by interfacial polarization and modifiers with different carbon chain lengths. Appl Phys Lett 112(16)
Yan Z, Zhang D, Zhou XF, Qi H, Luo H, Zhou KC, Abrahams I, Yan HX (2019) Silver niobate based lead-free ceramics with high energy storage density. J Mater Chem A 7(17):10702–10711
Zhou XF, Qi H, Yan ZN, Xue GL, Luo H, Zhang D (2019) Superior thermal stability of high energy density and power density in domain-engineered Bi0.5Na0.5TiO3–NaTaO3 relaxor ferroelectrics. ACS Appl Mater Interfaces 11(46):43107–43115
Chen X, Han X, Shen Q (2017) PVDF-based ferroelectric polymers in modern flexible electronics. Adv Electron Mater 3(5):1600460
Qian C, Zhu T, Zheng W, Bei R, Liu S, Yu D, Chi Z, Zhang Y, Xu J (2019) Improving dielectric properties and thermostability of CaCu3Ti4O12/polyimide composites by employing surface hydroxylated CaCu3Ti4O12 particles. ACS Appl Polym Mater 1(6):1263–1271
Liu XJ, Zheng MS, Chen G, Dang ZM, Zha JW (2022) High-temperature polyimide dielectric materials for energy storage: theory, design, preparation and properties. Energy Environ Sci 15(1):56–81
Li B, Polizos G, Manias E (2022) Interfacial effects on the dielectric properties of elastomer composites and nanocomposites//Dynamics of Composite Materials. Springer International Publishing, Cham, pp 225–249
Li B, Randall CA, Manias E (2022) Polarization mechanism underlying strongly enhanced dielectric permittivity in polymer composites with conductive fillers. J Phys Chem C 126(17):7596–7604
Yuan MX, Li B, Zhang S, Rajagopalan R, Lanagan MT (2020) High-Field dielectric properties of oriented poly (vinylidene fluoride-co-hexafluoropropylene): structure–dielectric property relationship and implications for energy storage applications. ACS Appl Polym Mater 2(3):1356–1368
Wang ZD, Wang XZ, Wang SL, He JY, Zhang T, Wang J, Wu GL (2021) Simultaneously enhanced thermal conductivity and dielectric breakdown strength in sandwich AlN/epoxy composites. Nanomaterials 11(8):1898
Wang ZD, Meng GD, Wang LL, Tian LL, Chen SY, Wu GL, Kong B, Cheng YH (2021) Simultaneously enhanced dielectric properties and through-plane thermal conductivity of epoxy composites with alumina and boron nitride nanosheets. Sci Rep 11(1):2495
Wang LL, Yang CX, Wang XY, Shen JY, Sun WJ, Wang JK, Yang GQ, Cheng YH, Wang ZD (2022) Advances in polymers and composite dielectrics for thermal transport and high-temperature applications. Compos A Appl Sci Manuf 107320
Jia LC, Jin YF, Ren JW, Zhao LH, Yan DX, Li ZM (2021) Highly thermally conductive liquid metal-based composites with superior thermostability for thermal management. J Mater Chem C 9(8):2904–2911
Zhao LH, Wei C, M, Ren JW, Li YC, Zheng JJ, Jia LC, Wang Z, Jia SL, (2022) Biomimetic nacreous composite films toward multipurpose application structured by aramid nanofibers and edge-hydroxylated boron nitride nanosheets. Ind Eng Chem Res 61(25):8881–8894
Zhang D, Liu WW, Guo R, Zhou KC, Luo H (2018) High discharge energy density at low electric field using an aligned titanium dioxide/lead zirconate titanate nanowire array. Adv Sci 5(2):1700512
Lu X, Deng W, Wei JD, Zhu YS, Ren PR, Wan YH, Yan FX, Jin L, Zhang L, Cheng ZY (2021) Filler size effects on the microstructure and properties of polymer-ceramic nanocomposites using a semicrystalline matrix. J Mater Sci 56:19983–19995
Li B, Sarkarat M, Baker A, Randall CA, Manias E (2021) Interfacial effects on the dielectric properties of elastomer/carbon-black/ceramic composites. MRS Advances 6:247–251
Yuan MX, Zhang G, Li B, Chung TCM, Rajagopalan R, Lanagan MT (2020) Thermally stable low-loss polymer dielectrics enabled by attaching cross-linkable antioxidant to polypropylene. ACS Appl Mater Interfaces 12(12):14154–14164
Funding
The authors gratefully acknowledge the financial supports from the National Natural Science Foundation of China (Nos. 52277028, 51903207), Shaanxi Provincial Natural Science Foundation of China (No.2022JM-186, 2021JQ-566), acknowledge the Analytic Instrumentation Center of XUST.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflicts of interests
The authors declare no conflict of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Cao, J., Zhou, W., Li, T. et al. Interfacial engineering of PZT/PVDF composites via insulating MgO as an interlayer towards enhanced dielectric performances. J Polym Res 30, 281 (2023). https://doi.org/10.1007/s10965-023-03655-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-023-03655-0