
Abstract
Vegetable oils, as raw materials for polyurethane foam, have attracted much attention for being non-toxic, biodegradable, renewable, and so on. However, polyurethane foam burns extremely easily and the vegetable oil grafted phosphorus/silicon flame retardant polyols alone do have insufficient fire-retardant efficiency. Herein, a new bio-based flame retardant (Polyol-P) was successfully synthesized through a ring-opening reaction by using epoxy soybean oil and phenylphosphonic acid as raw materials. Then, Polyol-P was first combined with melamine-formaldehyde resin-microencapsulated ammonium polyphosphate (MAPP) to modify polyurethane foam. The synergistic flame-retardant effects of Polyol-P with MAPP in polyurethane foam have been studied by thermogravimetric analysis, limiting oxygen index test, vertical burning test, and cone calorimeter. TG test results showed the addition of Polyol-P and MAPP promoted the degradation of polyurethane foam composites and the char residue rate at 800 °C of PU-P/15MAPP was significantly enhanced. Flame retardant test results confirmed PU-P/15MAPP exhibited LOI of 25.8% and passed the V-0 rating in the UL-94 test. Cone calorimeter test results indicated the peak heat release rate of PU-P and PU-P/15MAPP reduced from 395.10 kW/m2 to 291.92 kW/m2 and 222.72 kW/m2 compared with PU-0. Furthermore, the ratio of CO and CO2 remarkably reduced and the residual carbon yield of PU-P/15MAPP was the highest, and its value was 20.0 wt%. SEM confirmed that the char residue of PU-P/15MAPP was wrinkled and uneven, which was caused by the release of many volatile gases. FT-IR verified the formed structure of the phosphorous-containing carbonaceous structure. These results suggest that Polyol-P and MAPP have a good synergistic flame retardancy on polyurethane composites and can reduce fire risk.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polyurethanes (PUs) are a variety of polymeric substances containing urethane linkages (-NH-COO-), which consist of isocyanate, polyol, catalysts, surfactants, and so on [1, 2]. Due to high flexibility in structural design, polyurethanes have been fabricated into elastomers, adhesives, coatings, foams, and fibres [3, 4]. Among these materials, rigid polyurethane foam has been applied as thermal insulation in high-rise buildings because of its low density, low thermal conductivity, and high specific strength [5, 6]. However, owing to the porous structure and organic skeleton of polyurethane foam [7], it burns extremely easily and releases high heat and harmful gases, resulting in serious casualties and property losses. Consequently, enhancing the flame retardancy of polyurethanes has become an urgent problem to be solved in the industry [8].
With the popularization of the environmental protection concept and the increasing demand for sustainable development of resources, people gradually turn their targets to biomass resources [4, 9]. Because of their abundant resources, non-toxicity, renewability, and non-pollution characteristics [6, 10], biomass resources, such as starch [11], lignin [12, 13], chitosan [14,15,16], rice husk [17, 18], and sugar beet leaves [19], are flame retardant materials that meet the requirements of green development. Among many biomass resources, vegetable oils are made up generally of triglyceride molecules with varying degrees of unsaturation [20], which are applied in fuels (bio-diesel), polymers, lubricants, surfactants, and plasticizers [21]. There are many kinds of vegetable oils, including soybean, castor, sunflower, canola, and so on, which contain ester groups and carbon-carbon double bonds to provide modification sites for chemical modification [22]. However, vegetable oils have poor heat resistance and poor flame-retardant effect, and therefore flame-retardant modification of vegetable oils has become a current research hotspot.
Castor oil containing phosphorus polyols (COFPL) was synthesized through a multi-step reaction using castor oil, glycerol, and diethyl phosphate. And the author investigated the effect of COFPL on the properties of polyurethane foams [23]. The results showed that COFPL enhanced the thermal stability of polyurethane and the value of LOI reached 23.1% with the addition of 75 phr COFPL. Heinen et al. synthesized phosphorylated soybean oil polyol and used it as a raw material to synthesize flame-retardant polyurethane. When all of them replaced petroleum-based polyols, the LOI value was 21.8% [24]. Zhou et al. modified polyurethane foam containing 80 wt% PTOP. And the LOI value was 24.0% [25]. Aynur et al. carried out silylation of epoxidized soybean oil (ESO) by epoxy ring opening reaction of methoxy and ethoxy silane compounds, which were used to prepare polyurethanes labeled as PUSiTh and PUSiAm [26]. And the LOI values were 23.6% and 22.8%, respectively. Whereas, the above vegetable oil grafted phosphorus/silicon flame retardant polyols alone do have insufficient fire-retardant efficiency and the LOI of flame-retardant polyurethane foams is hard to over 24%. The joint use of disparate flame retardants is proposed to assemble a multiform synergistic device to satisfy the need in the actual production [27].
Inorganic flame retardants, such as ammonium polyphosphate (APP), Al(OH)3, Mg(OH)2, and so on, are easy to obtain and low cost [28, 29]. As a common flame retardant for high nitrogen and phosphorus elements, APP is added with additives. However, APP has poor compatibility with organic polymer materials, which may seriously reduce flame-retardant efficiency [30]. Microencapsulation technology can solve the defect of inorganic flame-retardant [31]. Whereas, there are few reports on the combination of synergistic flame retardancy for the formulation of soybean oil phosphate-based polyols and modified ammonium polyphosphate.
In this paper, a new soybean oil-based flame retardant was synthesized from epoxidized soybean oil and phenylphosphonic acid and compounded with modified ammonium polyphosphate to prepare flame retardant polyurethane foams. The thermal stability, flame retardancy, morphological characteristics, and mechanical properties of polyurethane were evaluated using thermogravimetric analyser, cone calorimeter, mechanical property tests, and scanning electron microscopy. The synergistic effect between Polyol-P and MAPP improves the flame retardancy and mechanical properties of polyurethane foam.
Experimental
Materials
Epoxidized soybean oil (ESO) with an epoxide index of 3.80 mol/kg was supplied by Aladdin Co., Ltd. (Shanghai China). Phenyl phosphonic acid (PPA) was purchased from Aladdin Co., Ltd. (Shanghai China). Tertiary butanol and n-hexane were received from Shanghai Lingfeng Chemical Reagents Co., Ltd. (Shanghai China). Melamine-formaldehyde resin‑microencapsulated ammonium polyphosphate (MAPP) was provided by Guangzhou Yinyuan New Material Co. Ltd. (Guangzhou China). The petrochemical polyol (PPG 4110), with a hydroxyl value of 440 mg KOH/g, was supplied by Xuzhou Yahiya New Material Co. LTD. Polymethylene polyphenyl isocyanate (PM-200), with an NCO content of 30 wt%, was bought by Jining Huakai Resin Co., Ltd. (Jining China). Dibutyltin dilaurate (DBTDL) and triethanolamine (TEOA), as catalysers 1 and 2, were provided by Aladdin Co., Ltd. (Shanghai China). Silicon surfactant (AK 8805) was provided by Jiangsu Maysta Chemical Co., Ltd. (Nanjing China). Distilled water was used as the chemical blowing agent.
Synthesis of Polyol-P
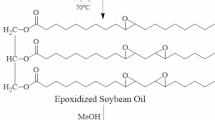
The procedure was inspired by those works of literature [32, 33]. 50 g of ESO and 25 g of tertiary butanol were added to a 250 mL four-neck flask equipped with a mechanical stirrer, a refluxing column, a thermometer, and a dropping funnel. The reaction device was placed in an oil bath of 85 °C. Then, the mixture of phenyl phosphonic acid (12.5 g) and tertiary butanol (25 g) was dropped into the flask for 25 min. The reaction mixture was stirred at 85 °C for 9 h. After the reaction, the solvent was removed using a rotary evaporator. A soy-based polyol labelled Polyol-P was obtained. The hydroxyl value of Polyol-P was 195 mg KOH/g. Although Polyol-P can consider a mixture of different related polyols, a certain polyol is listed in Fig. 1 and it does not hinder the modification of polyurethane foam [33]. The result further facilitates the practical application of the method.

The synthesis route of Polyol-P
Preparation of PU composites
Table 1 depicted the formulation of PU composites. The value of R (R=-NCO/-OH) of PU composites was 1.2. First, polyol, water, n-hexane, dibutyltin dilaurate (DBTDL), triethanolamine (TEOA), AK8805, and MAPP were thoroughly mixed in a plastic beaker at 1000 rpm for 10 min to form a uniform mixture. And then PM-200 was immediately added to the mixture and stirred (1500 rpm) for 10 s. The mixture was freely foamed in the plastic cup and cured at room temperature for 3 days. Finally, PU composites to be tested were cut into standard-size pieces.
Methods
The infrared spectra were analysed by NEXUS670 (Thermo Nicolet Corporation, US) within the wave number range of 4000 − 400 cm− 1. The 1H NMR and 31P NMR of products were carried out on a Bruker Avance III HD-400 spectrometer using CDCl3 as the solvent. The surface morphology of PU composites and the residual carbon after cone calorimeter tests were determined by JSM-5610LV scanning electron microscopy. To improve the electrical conductivity, all specimens were sprayed with gold coating. The apparent density of PU composites was measured according to ASTM D1622, with a sample size of 30 × 30 × 30 mm3. The compressive strength at 10% was examined by a universal tensile instrument CMT4000 according to ASTM D 1621, and the dimension of the sample was 50 mm × 50 mm × 25 mm. Thermal degradation behaviour of PU composites was obtained from a NETZSCH TG 209 F1 apparatus at the heating rate of 15 °C/min from 30 to 800 °C under N2 atmosphere. LOI values of samples were detected via a JF-3 oxygen index meter in line with ASTM D2863-97, with the dimensions of 100 mm × 10 mm × 10 mm. The vertical burning test was performed by a JF-3 instrument according to ASTM D3801-2010, with a sample size of 130 mm× 10.0 mm × 10.0 mm. The combustion behaviour of PU composites was characterized by cone calorimetry (Fire Testing Technology) according to ISO 5660. Each specimen with the size of 100 mm × 100 mm × 25 mm was exposed horizontally to 35 kW/m2 external heat flux.
Results and discussion
Characterization of Polyol-P
Figure 2 depicted the 1H NMR spectra of ESO and Polyol-P. As shown in 1H NMR of ESO, the peak at 0.8 ppm was matched with the protons of -CH3 (labelled a), and the signals at 5.26 ppm and 4.13–4.31 ppm were allocated to the protons of the methylene protons of triglyceride structure (labelled i and h). The peak at 2.9–3.1 ppm was associated with protons of the epoxy groups (labelled f). Whereas, through the comparison of 1H NMR between ESO and Polyol-P, we could see that the peak at 2.9–3.1 ppm (labelled f) disappeared and the multi-peaks around 7.46–7.80 ppm (labelled j) corresponding to protons of benzene skeleton were observed. These results suggested reaction occurred between epoxidized soybean oil and PPA.

1H NMR of ESO and Polyol-P
31P NMR spectra of Polyol-P and PPA were measured and the results were shown in Fig. 3. As can be seen, there was a sharp signal at 34.06 ppm in the 31P NMR spectra of Polyol-P. And the peak of PPA was observed at 15.82 ppm. From the above analysis, it could be concluded that Polyol-P was synthesized successfully.

31P NMR spectra of Polyol-P and PPA
FT-IR spectra of ESO and Polyol-P were depicted in Fig. 4. As shown in the FT-IR spectrum of ESO, the peaks at 2927 cm− 1 and 2858 cm− 1 were attributed to the antisymmetric stretching vibration and symmetric stretching vibration of -CH3. Furthermore, the absorption peaks at 1740 cm− 1 and 826 cm− 1 were ascribed to the stretching vibration of the C = O and epoxy groups, respectively [34]. In contrast, the FT-IR spectra of Polyol-P showed a strong and broad peak at 3424 cm− 1 assigned to -OH, while the peak of the epoxy group of ESO at 826 cm− 1 disappeared. And the absorption peaks at 1438 cm− 1, 1157 cm− 1, and 750 cm− 1 corresponded to stretching vibration of the benzene skeleton, P = O, and P-C, respectively [33]. These results implied that the phosphorous-phenyl structure was introduced into Polyol-P.

FT-IR spectra of ESO and Polyol-P
Thermal performance of PU composites
As a research instrument on pyrolysis behaviour, thermogravimetric analyser can provide relevant information such as thermal stability, degradation behaviour, char formation, and so on [35,36,37]. Herein, the thermal stability of PU composites was investigated by thermogravimetry under N2 atmosphere. The TG and derivative TG (DTG) curves of PU composites were depicted in Fig. 5. The selected data were as follows: temperature of 5% weight loss (T5%), the temperature of the maximum degradation rate (Tmax), and the char residue at 800 °C, which were summarized in Table 2 [8]. All samples showed two weight-loss stages coinciding with results in the literature [27, 37]. The first stage was at 200–385 °C. This could be ascribed to the broken of the urethane bond and it was further decomposed into diimine carbides, olefin, aldehydes, ketones, carbon dioxide, and water [31, 38].

The thermal performance of PU composites: A TGA and B DTG
The second stage at 385–500 °C was attributed mainly to the decomposition of polyols [39]. As shown in Fig. 5A, T5% of PU-0 was 235 °C and Tmax was 322 °C and 470 °C, respectively. The char residue of PU-0 was 15.1 wt% when heated to 800 °C. When added Polyol-P, T5% and Tmax1 of PU-P dropped to 193 °C and 312 °C. This may be explained by the first reason that urethane linkages of polyurethane foam are formed by secondary OH groups with isocyanate groups [40, 41]. The second reason is that the energy of the P-O bond (149 kJ/mol) is much lower than the C-O bond (256 kJ/mol). When MAPP was added, T5% and Tmax of PU-P/5MAPP, PU-P/10MAPP, and PU-P/15MAPP continued to decrease. These results mainly resulted from that polyphosphoric acid generated by the thermal decomposition of MAPP promoted decomposition of polyurethane matrix [31]. Furthermore, PU-P, PU-P/5MAPP, PU-P/10MAPP, and PU-P/15MAPP exhibited char residues of 23.6 wt%, 17.4 wt%, 21.9 wt%, and 28.1 wt%, respectively, which were higher than that of PU-0. Although the decomposition temperature of polyurethane composites reduced, polyphosphate produced by the early decomposition of Polyol-P and MAPP is conducive to the formation of a stable and dense carbon layer on the surface, which can well block the external heat and oxygen generation [27, 42].
Flame retardancy tests of PU composites
To assess the flammability of PU composites, the LOI and UL-94 test results of PU composites were listed in Table 3. PU-0 possessed the LOI value of 20.0% and failed in the UL-94 tests, which also observed a molten dripping phenomenon. The high flammability of PU-0 was attributed to the large surface area and porous structure of polyurethane [43, 44]. However, when Polyol-P was added to the material, the LOI of PU-P increased to 22.7% and passed the V-1 rating in the vertical burning test. When MAPP was added to polyurethane foam, the LOI values of PU-P/MAPP increased continuously. When the addition of MAPP ranged from 5 wt% to 15 wt%, the LOI value of PU-P/MAPP increased from 23.5% to 25.8%.
The digital images of the UL-94 test of PU composites were given in Fig. 6. It was noteworthy that the burning area of PU-0 reduced drastically while the burned PU-P/MAPP retained most of the residual. This phenomenon was observed mainly because MAPP and Polyol-P released PO* and PO2*radicals to quench the H* and HO* and terminated the free radical reaction [30]. Meanwhile, polyphosphoric produced by MAPP can catalyse the forming of a dense carbon layer to hinder the energy exchange in the combustion zone [43,44,45], thereby effectively improving the flame retardancy of polyurethane. By combining all analysis results, the incorporation of Polyol-P combination with MAPP jointly has a good synergistic flame-retardant effect.

Digital photos of UL-94 test of PU-0 (A, A'), PU-P (B, B'), PU-P/5MAPP (C, C'), PU-P/10MAPP (D, D'), and PU-P/15MAPP (E, E')
Combustion behaviour of PU composite
Cone calorimeter test is used to evaluate the combustion behavior of polyurethane materials in a real fire environment [46]. It was listed the curves of the heat release rate (HRR) and the mass change of polyurethane composites in Fig. 7.

A HRR and B Mass loss curves of PU-0, PU-P, and PU-P/15MAPP
Relevant characteristic parameters, such as ignition time (TTI), total heat release (THR), peak heat release rate (PHRR), and ignition performance index (FPI), were shown in Table 4. As can be seen in Table 4, the TTI (2–4 s) of all samples was very short, which was attributed to the porous structure and large surface area of polyurethane [43, 44]. It is well known that HRR and THR are important parameters for studying flammability, which can be used to express fire intensity and fire spread rate [46]. From Fig. 7A, it was clear that PU-0 achieved the maximum value of HRR (395.1 kW/m2) after being ignited for 18 s. With the addition of Polyol-P, the PHRR of PU-P decreased to 291.9 kW/m2. Furthermore, when incorporating MAPP and Polyol-P, the PHRR jointly further decreased to 222.7 kW/m2 and reduced by 43.6% than PU-0. This result suggested that the combination of MAPP with Polyol-P had a significant inhibitory effect on the combustion intensity of polyurethane composites. In addition, it was worth noting that the reduction of THR of PU composites was not obvious and the THR value of PU-P/15MAPP bounced. This phenomenon was related to more vapour-phase products released from Polyol-P and MAPP due to the gaseous phase flame retardancy [47, 48].
CO and CO2 are two representative gases used to investigate toxic gas emissions of polyurethane composites [49]. The ratio of CO and CO2 was shown in Table 4. With the addition of Polyol-P and MAPP, the ratio of CO and CO2 decreased continuously, further revealing the effective smoke suppression of Polyol-P and MAPP for polyurethane [50]. The degree of the volatile gas combustion is reflected by the average effective heat combustion (Av-EHC) in the gas phase. The Av-EHC of PU-P was 22.63 MJ/kg, which was 20.4% lower than that of PU-0, indicating a quenching effect of Polyol-P. When the additional amount of MAPP was 15 wt%, the value of Av-EHC increased to 26.07 MJ/kg, which suggested MAPP enhanced the flame-retardant effect of the condensed phase.
The mass loss curves disclose the effect of char residue on combustion-restraining behavior during the fire [3]. As plotted in Fig. 7B, the char yield of PU-0 was 0.8 wt%, whereas the residues of PU-P and PU-P/15MAPP reached 12.3 wt% and 20.0 wt%, respectively. This result was caused by the fact that the charring of the substrate was promoted by the dehydration of phosphate derivatives produced by Polyol-P and MAPP in the condensed phase [37]. A-MLR represents the thermal cracking, volatilization, and combustion of materials under a certain fire intensity [50]. The value of A-MLR dropped from 0.051 g/s to 0.041 g/s from Table 4, indicating that much more char residues were formed (confirmed by Fig. 7B). There is a good synergistic flame-retardant effect of Polyol-P combined with MAPP.
Finally, fire hazards are analysed by calculating the fire performance index (FPI), which is the ratio of TTI and PHRR [51]. A higher FPI value indicates lower fire risk. Compared to PU-0, the FPI values of PU-P and PU-P/15MAPP increased to 0.014 m2s/kW and 0.018 m2s/kW, respectively. These results suggest that MAPP and Polyol-P endow PU composites with higher fire safety.
Char residue analysis and flame-retardant mechanism
Firstly, the morphologies of the residues were studied through digital photos and SEM micrographs. The digital photos of residual chars were described in Fig. 8. The residual carbon layer of PU-0 (Fig. 8A) was light and would be easily broken, which meant that PU-0 completely disintegrated during the combustion process [52]. In Fig. 8B, Polyol-P could form a relatively dense carbonaceous layer. With the addition of MAPP, the carbon residue rate of PU-P/15MAPP increased significantly and the densification of the char residue was improved (Fig. 8C).

Digital photographs of the residues after the cone calorimeter test. A PU-0, B PU-P, and C PU-P/15MAPP
SEM images of the residues were shown in Fig. 9. It was clear that the char structure of PU-0 was broken and loose [53]. Whereas, the char of PU-P was observed to be compact, smooth, and wrinkled, which could effectively reduce the release of heat [46]. The coke of PU-P/15MAPP was creased and uneven, with obvious pores on the surface. This resulted from many volatile gases released by MAPP, which increased the combustion intensity [54]. This result was consistent with the analysis of cone calorimeter test.

SEM images of residues after cone calorimeter test: A PU-0, B PU-P, and C PU-P/15MAPP
Analysis of the residue composition contributed to revealing the fire-retardant mechanism [43]. The FTIR spectra of PU-0, PU-P, and PU-P/15MAPP residual chars were shown in Fig. 10. The peak at 3435 cm− 1 corresponded to the stretching vibration of N-H [6]. Peaks around 2925 − 2850 cm− 1 were typical for the stretching vibration of -CH2-. And The absorption peaks at 1450 cm− 1, 1370 cm− 1, and 753 cm− 1 could be attributed to the vibration adsorption of -CH3 and CAr-H, respectively. The patterns of PU-P and PU-P/15MAPP composites were similar. Compared with PU-0, the characteristic absorption of P = O, P-O-C, and P-O-P near 1214 cm− 1, 1068 cm− 1, and 981 cm− 1 were observed in the FTIR of PU-P and PU-P/15MAPP. And the absorption intensity of P-O-P of PU-P/15MAPP was stronger than that of PU-P. The result suggested phosphoric acids interacted with the polyurethane matrix and formed phosphorus-rich residues [37]. The result of the FTIR spectrum showed that the formed phosphorous-containing carbonaceous structure contributed to the enhancement of fire retardancy.

FT-IR spectra of the residual char of PU-0, PU-P, and PU-P/15MAPP
Summarizing the abovementioned results, the synergistic flame-retardant mechanism of the Polyol-P/MAPP system applied in PU composites is obtained, and is depicted in Fig. 11. When Polyol-P was alone added to PU composites, the flame-retardant mechanism was dominated by the gas phase. This result was consistent with the average effective heat combustion (Av-EHC), which reflected the degree of the volatile gas combustion. The Av-EHC of PU-P was 22.63 MJ/kg, which was 20.4% lower than that of PU-0, indicating a quenching effect of Polyol-P. Polyol-P could produce PO* and PO2* free radicals capturing *OH and H* active radicals, thus blocking the combustion reaction. Meanwhile, when MAPP was added to the PU-P, the decomposition of NH3 and water vapor produced by MAPP could dilute combustible gases and remove some heat. The value of Av-EHC increased to 26.07 MJ/kg, which suggested MAPP enhanced the flame-retardant effect of the condensed phase. Polyphosphoric acid produced by the thermal decomposition of MAPP and Polyol-P could dehydrate and carbonize polyurethane molecular chain to form a condensed carbon layer, which inhibited mass and heat transmission in a fire. In conclusion, the synergistic action of the gas-phase and condensed-phase flame-retardant mechanism significantly enhances the fire-retardant ability of PU composites.

Possible flame-retardant mechanism of the PU composites incorporated with Polyol-P and MAPP
Cell morphology of PU composites
The mechanical property of PU is observably affected by cell morphology [55]. Therefore, SEM images with 50 times magnification of each cross section from PU composites were shown in Fig. 12. It was obvious that all samples were composed of spherical and polyhedron cells [56]. The surface of PU-0 and PU-P exhibited a relatively clean and smooth morphology at high magnification. When 5 wt% MAPP was added, the cellular structure of PU-P/5MAPP was relatively uniform. This indicated MAPP particles were evenly dispersed, with good compatibility with polyurethane foam. However, when 10 wt% MAPP and 15 wt% MAPP were added to polyurethane foam, the surfaces became much rougher. In addition, the rupture of some struts and the collapse of the cellular structure were observed, with particles agglomerating. This may be due to the limited compatibility between polyurethane and MAPP.
As shown in Fig. 12A, the average cell size of PU-0 was 191.27 μm, with a cell size distribution of 125–275 μm. With the addition of Polyol-P, the average cell size of PU-P was increased to 223.23 μm, with a cell size distribution of 175–280 μm (Fig. 12B). The reason was that the hydroxyl value of.

SEM and cell sizes distribution of PU composites: A PU-0, B PU-P, C PU-P/5MAPP, D PU-P/10MAPP, and E PU-P/15MAPP
Polyol-P was lower than PPG 4110, resulting in a decrease in crosslink density. When MAPP was added to polyurethane foam, the average cell size became smaller. The cell size of PU-P/10MAPP was the smallest (103.32 μm) and the cell-size distribution was 40–180 μm (Fig. 12D). However, the average cell size of PU/15MAPP increased to 105.55 μm, and the cell-size distribution expanded to 70–180 μm (Fig. 12E). These results indicated MAPP possessed a certain nucleation effect, which effectively reduced the free energy for the nucleation during foaming [4, 57]. In addition, the incorporation of MAPP increased the viscosity of the mixture, preventing the merging of air bubbles, which could lead to a reduction in cell size [55].
Mechanical properties of PU composites
Apparent density and compressive strength are important physical properties of polyurethane, which directly affect the application of polyurethane composites [58, 59]. Figure 13 showed the density and compressive of PU composites. As can be seen from Fig. 13A, PU-0 possessed a density of 47.0 kg/m3. With the addition of Polyol-P, the density of PU-P increased to 66.0 kg/m3, which was ascribed to increasing the viscosity of the reaction mixture. Furthermore, the foam density increased from 68.9 kg/m3 to 76.2 kg/m3 further as the amount of MAPP was added.

A apparent density and B compression strength of PU composites
This was because MAPP was a solid filler, which had a higher density [60]. It was noteworthy that the compressive strength of the polyurethane increased from 0.130 MPa to 0.260 MPa with the addition of Polyol-P and MAPP (Fig. 13B), which could be ascribed to the high density, resulting in the high compressive strength [61]. Apart from density, the compressive strength of polyurethane depends on the porous structure of the polymer [61]. The addition of MAPP makes cell size decrease and cell number increase, leading to the fact that more cell walls per unit area of the foam encountered more applied load.
Conclusion
In this paper, a new bio-based flame-retardant Polyol-P was combined with MAPP to modify polyurethane foam. The test results showed that the joint addition of Polyol-P and MAPP enhanced the flame retardancy of polyurethane. Compared with PU-0, the LOI value of PU-P/15MAPP increased from 20.0% to 25.8% and passed the V-0 rating in the vertical burning test. In addition, cone calorimetry test results showed that the total heat release of PU-P/15MAPP significantly increased. Meanwhile, compared with PU-0, the peak heat release rate of PU-P and PU-P/15MAPP reduced from 395.10 kW/m2 to 291.92 kW/m2 and 222.72 kW/m2 respectively. The flame-retardant mechanism indicated that Polyol-P and MAPP produced active free radicals capturing *OH and H* active radicals, thus blocking the combustion reaction. At the same time, Polyol-P and MAPP jointly promoted the generation of the cross-linking system, forming more protective carbon layers gases and removing some heat. Therefore, the synergistic action of the gas-phase and condensed-phase flame-retardant mechanism significantly enhanced the fire-retardant ability of PU composites. Additionally, the addition of MAPP induced a significant increase in compressive strength, and the maximum compressive strength of PU-P/15MAPP was 0.260 MPa, compared to PU-0. These results indicate that the PU-P/15MAPP system improved the mechanical properties and heat resistance of polyurethane foam more effectively than the PU-P system.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Pillai PKS, Li S, Bouzidi L, Narine SS (2016) Metathesized palm oil polyol for the preparation of improved bio-based rigid and flexible polyurethane foams. Ind Crop Prod 83:568–576. https://doi.org/10.1016/j.indcrop.2015.12.068
Wang S, Qian L, Xin FT (2018) The synergistic flame-retardant behaviors of pentaerythritol phosphate and expandable graphite in rigid polyurethane foams. Polym Compos 39:329–336. https://doi.org/10.1002/pc.23939
Yan J, Xu P, Zhang P, Fan H (2021) Surface-modified ammonium polyphosphate for flame-retardant and reinforced polyurethane composites. Colloid Surf A 626:127092–127102. https://doi.org/10.1016/j.colsurfa.2021.127092
Zhang B, Feng Z, Yang Y, Xu X, Chen D, Huang X, Liu C, Liu X, Tang G (2022) Facile synthesis of melamine phytates and its application in rigid polyurethane foam composites targets for improving fire safety. Plast Rubber Compos 2022:1–15. https://doi.org/10.1080/14658011.2021.2024647
Prociak A, Kurańska M, Uram K, Wójtowicz M (2021) Bio-polyurethane foams modified with a mixture of bio-polyols of different chemical structures. Polymers 13:2469–2478. https://doi.org/10.3390/polym13152469
Yang S, Zhang B, Liu M, Yang Y, Liu X, Chen D, Wang B, Tang G, Liu X (2021) Fire performance of piperazine phytate modified rigid polyurethane foam composites. Polym Advan Technol 32:4531–4546. https://doi.org/10.1002/pat.5454
Zhang B, Feng Z, Han X, Wang B, Yang S, Chen D, Peng J, Yang Y, Liu X, Tang G (2021) Effect of ammonium polyphosphate/cobalt phytate system on flame retardancy and smoke & toxicity suppression of rigid polyurethane foam composites. J Polym Res 28:407–422. https://doi.org/10.1007/s10965-021-02763-z
Liu DY, Zhao B, Wang JS, Liu PW, Liu YQ (2018) Flame retardation and thermal stability of novel phosphoramide/expandable graphite in rigid polyurethane foam. J Appl Polym Sci 135:46434–46443. https://doi.org/10.1002/app.46434
Liu X, Sui Y, Guo P, Chen R, Mu J (2022) A flame retardant containing biomass-based polydopamine for high-performance rigid polyurethane foam. New J Chem 46:11985–11993. https://doi.org/10.1039/d2nj00671e
Dowbysz A, Samsonowicz M, Kukfisz B (2022) Recent advances in bio-based additive flame retardants for thermosetting resins. Int J Environ Res Pub Health 19:4828–4853. https://doi.org/10.3390/ijerph19084828
Choi KW, Kim JM, Kwon TS, Kang SW, Song JI, Park YT (2021) Mechanically sustainable starch-based flame-retardant coatings on polyurethane foams. Polymers 13:1286–1296. https://doi.org/10.3390/polym13081286
Zhu H, Peng Z, Chen Y, Li G, Wang L, Tang Y, Pang R, Khan Z, Wan P (2014) Preparation and characterization of flame-retardant polyurethane foams containing phosphorus-nitrogen-functionalized lignin. RSC Adv 4:55271–55279. https://doi.org/10.1039/c4ra08429b
Wang Y, Zhang Y, Liu B, Zhao Q, Qi Y, Wang Y, Sun Z, Liu B, Zhang N, Hu W, Xie H (2020) A novel phosphorus-containing lignin-based flame retardant and its application in polyurethane. Compos Commun 21:100382–100385. https://doi.org/10.1016/j.coco.2020.100382
Lin B, Yuen ACY, Li A, Zhang Y, Chen TBY, Yu B, Lee EWM, Peng S, Yang W, Lu HD, Chan QN, Yeoh GH, Wang CH (2020) MXene/chitosan nanocoating for flexible polyurethane foam towards remarkable fire hazards reductions. J Hazard Mater 381:120952–102961. https://doi.org/10.1016/j.jhazmat.2019.120952
Wong EHH, Fan KW, Lei L, Wang C, Baena JC, Okoye H, Fam W, Zhou D, Oliver S, Khalid A, Yeoh GH, Wang CH, Boyer C (2021) Fire-resistant flexible polyurethane foams via nature-inspired chitosan-expandable graphite coatings. ACS Appl Polym Mater 3:4079–4087. https://doi.org/10.1021/acsapm.1c00580
Carosio F, Ghanadpour M, Alongi J, Wagberg L (2018) Layer-by-layer-assembled chitosan/phosphorylated cellulose nanofibrils as a bio-based and flame protecting nano-exoskeleton on PU foams. Carbohyd Polym 202:479–487. https://doi.org/10.1016/j.carbpol.2018.09.005
Attia NF, Saleh BK (2019) Novel synthesis of renewable and green flame-retardant, antibacterial and reinforcement material for styrene-butadiene rubber nanocomposites. J Therm Anal Calorim 139:1817–1827. https://doi.org/10.1007/s10973-019-08582-1
Attia NF, Afifi HA, Hassan MA (2015) Synergistic study of carbon nanotubes, rice husk ash and flame retardant materials on the flammability of polystyrene nanocomposites. Mater Today: Proc 2:3998–4005. https://doi.org/10.1016/j.matpr.2015.08.029
Attia NF (2022) Sustainable and efficient flame retardant materials for achieving high fire safety for polystyrene composites. J Therm Anal Calorim 147:5733–5742. https://doi.org/10.1007/s10973-021-10948-3
Ji D, Fang Z, He W, Luo Z, Jiang X, Wang T, Guo K (2015) Polyurethane rigid foams formed from different soy-based polyols by the ring opening of epoxidized soybean oil with methanol, phenol, and cyclohexanol. Ind Crop Prod 74:76–82. https://doi.org/10.1016/j.indcrop.2015.04.041
Guo Y, Hardesty JH, Mannari VM, Massingill JL (2007) Hydrolysis of epoxidized soybean oil in the presence of phosphoric acid. J Am Oil Chem Soc 84:929–935. https://doi.org/10.1007/s11746-007-1126-5
Fahmy HM, Amr A, Aly AA, Sayed SM (2019) Synthesis of castor oil/2,4-toluene diisocyanate adduct to impart water repellency and antibacterial properties for cotton/polyester fabric. J Coat Technol Res 16(1):31–39. https://doi.org/10.1007/s11998-018-0097-9
Zhang L, Zhang M, Hu L, Zhou Y (2014) Synthesis of rigid polyurethane foams with castor oil-based flame retardant polyols. Ind Crop Prod 52:380–388. https://doi.org/10.1016/j.indcrop.2013.10.043
Heinen M, Gerbase AE, Petzold CL (2014) Vegetable oil-based rigid polyurethanes and phosphorylated flame-retardants derived from epoxidized soybean oil. Polym Degrad Stab 108:76–86. https://doi.org/10.1016/j.polymdegradstab.2014.05.024
Zhou W, Bo C, Jia P, Zhou Y, Zhang M (2018) Effects of tung oil-based polyols on the thermal stability, flame retardancy, and mechanical properties of rigid polyurethane foam. Polymers 11:45–61. https://doi.org/10.3390/polym11010045
ÖZŞEker A, KaradenİZ K, ŞEn MY (2019) Silylation of epoxidized soybean oil with triethoxysilanes, synthesis and characterization of their polyurethanes. Turk J Chem 43:1365–1382. https://doi.org/10.3906/kim-1904-11
Zhang H, Lyu L, Huang Z, Yan Y (2022) Acoustic performance and flame retardancy of ammonium polyphosphate/diethyl ethylphosphonate rigid polyurethane foams. Polymers 14:420–437. https://doi.org/10.3390/polym14030420
Wang Z, Jiang Y, Yang X, Zhao J, Fu W, Wang N, Wang DY (2022) Surface modification of ammonium polyphosphate for enhancing flame-retardant Properties of thermoplastic polyurethane. Materials 15:1990–2002. https://doi.org/10.3390/ma15061990
Abdelmoaty A, Abou-okeil A, Hanna A, Amr A (2022) The flame retardancy of fabricated cotton incorporated with Mg(OH)2 nano-particles. Egypt J Chem 65. https://doi.org/10.21608/ejchem.2022.122202.5472
Feng Y, Wu W, Wang Z, Zhao T (2022) POSS-modified ammonium polyphosphate for improving flame retardant of epoxy resins. Polym Advan Technol 33:1190–1201. https://doi.org/10.1002/pat.5592
Yang Y, Chen W, Liu M, Zhu Q, Liu X, Zhang B, Chen D, Liu X, Zhang K, Tang G (2021) Flame retarded rigid polyurethane foam composites based on gel-silica microencapsulated ammonium polyphosphate. J Sol-Gel Sci Technol 98:212–223. https://doi.org/10.1007/s10971-021-05484-3
Chen MJ, Wang X, Tao MC, Liu XY, Liu ZG, Zhang Y, Zhao CS, Wang JS (2018) Full substitution of petroleum-based polyols by phosphorus-containing soy-based polyols for fabricating highly flame-retardant polyisocyanurate foams. Polym Degrad Stab 154:312–322. https://doi.org/10.1016/j.polymdegradstab.2018.07.001
Acuña P, Zhang J, Yin GZ, Liu XQ, Wang DY (2020) Bio-based rigid polyurethane foam from castor oil with excellent flame retardancy and high insulation capacity via cooperation with carbon-based materials. J Mater Sci 56:2684–2701. https://doi.org/10.1007/s10853-020-05125-0
de Haro JC, Lopez-Pedrajas D, Perez A, Rodriguez JF, Carmona M (2019) Synthesis of rigid polyurethane foams from phosphorylated biopolyols. Environ Sci Pollut Res 26:3174–3183. https://doi.org/10.1007/s11356-017-9765-z
Qian L, Li L, Chen Y, Xu B, Qiu B (2019) Quickly self-extinguishing flame retardant behavior of rigid polyurethane foams linked with phosphaphenanthrene groups. Compos Part B-Eng 175:107186–107195. https://doi.org/10.1016/j.compositesb.2019.107186
Chi Z, Guo Z, Xu Z, Zhang M, Li M, Shang L, Ao Y (2020) A DOPO-based phosphorus-nitrogen flame retardant bio-based epoxy resin from diphenolic acid: synthesis, flame-retardant behavior and mechanism. Polym Degrad Stab 176:109151–109164. https://doi.org/10.1016/j.polymdegradstab.2020.109151
Attia NF, Mohamed GG, Ismail MM, Abdou TT (2020) Influence of organic modifier structures of 2D clay layers on thermal stability, flammability and mechanical properties of their rubber nanocomposites. J Nanostruct Chem 10:161–168. https://doi.org/10.1007/s40097-020-00338-w
Tang G, Jiang H, Yang Y, Chen D, Liu C, Zhang P, Zhou L, Huang X, Zhang H, Liu X (2020) Preparation of melamine-formaldehyde resin-microencapsulated ammonium polyphosphate and its application in flame retardant rigid polyurethane foam composites. J Polym Res 27:375–389. https://doi.org/10.1007/s10965-020-02343-7
Chan YY, Ma C, Zhou F, Hu Y, Schartel B (2021) Flame retardant flexible polyurethane foams based on phosphorous soybean-oil polyol and expandable graphite. Polym Degrad Stab 191:109656–109671. https://doi.org/10.1016/j.polymdegradstab.2021.109656
Stirna U, Fridrihsone A, Lazdiņa B, Misāne M, Vilsone D (2012) Biobased polyurethanes from rapeseed oil polyols: structure, mechanical and thermal properties. J Polym Environ 21:952–962. https://doi.org/10.1007/s10924-012-0560-0
Xu J, Wu Y, Zhang B, Zhang G (2020) Synthesis and synergistic flame-retardant effects of rigid polyurethane foams used reactive DOPO‐based polyols combination with expandable graphite. J Appl Polym Sci 138:50223–50232. https://doi.org/10.1002/app.50223
Yang S, Liu X, Tang G, Long H, Wang B, Zhang H, Ji Y, Yang Y (2021) Fire retarded polyurethane foam composites based on steel slag/ammonium polyphosphate system: a novel strategy for utilization of metallurgical solid waste. Polym Adv Technol 33:452–463. https://doi.org/10.1002/app.50223
Pang XY, Xin YP, Shi XZ, Xu JZ (2019) Effect of different size-modified expandable graphite and ammonium polyphosphate on the flame retardancy, thermal stability, physical, and mechanical properties of rigid polyurethane foam. Polym Eng Sci 59:1381–1394. https://doi.org/10.1002/pen.25123
Cheng J, Niu S, Ma D, Zhou Y, Zhang F, Qu W, Wang D, Li S, Zhang X, Chen X (2020) Effects of ammonium polyphosphate microencapsulated on flame retardant and mechanical properties of the rigid polyurethane foam. J Appl Polym Sci 137:49591–49598. https://doi.org/10.1002/app.49591
Wang SX, Zhao HB, Rao WH, Huang SC, Wang T, Liao W, Wang YZ (2018) Inherently flame-retardant rigid polyurethane foams with excellent thermal insulation and mechanical properties. Polymer 153:616–625. https://doi.org/10.1016/j.polymer.2018.08.068
Wang C, Wu Y, Li Y, Shao Q, Yan X, Han C, Wang Z, Liu Z, Guo Z (2018) Flame-retardant rigid polyurethane foam with a phosphorus-nitrogen single intumescent flame retardant. Polym Adv Technol 29:668–676. https://doi.org/10.1002/pat.4105
Rao WH, Hu ZY, Xu HX, Xu YJ, Qi M, Liao W, Xu S, Wang YZ (2017) Flame-retardant flexible polyurethane foams with highly efficient melamine salt. Ind Eng Chem Res 56:7112–7119. https://doi.org/10.1021/acs.iecr.7b01335
Kang AH, Shang K, Ye DD, Wang YT, Wang H, Zhu ZM, Liao W, Xu SM, Wang YZ, Schiraldi DA (2017) Rejuvenated fly ash in poly (vinyl alcohol)-based composite aerogels with high fire safety and smoke suppression. Chem Eng J 327:992–999. https://doi.org/10.1016/j.cej.2017.06.158
Yin Z, Lu J, Yu X, Jia P, Tang G, Zhou X, Lu T, Guo L, Wang B, Song L, Hu Y (2021) Construction of a core-shell structure compound: ammonium polyphosphate wrapped by rare earth compound to achieve superior smoke and toxic gases suppression for flame retardant flexible polyurethane foam composites. Compos Commun 28:100939–100344. https://doi.org/10.1016/j.coco.2021.100939
Nomura K, Terwilliger P (2019) Self-dual leonard pairs. Spec Matrices 7:1–19. https://doi.org/10.1515/spma-2019-0001
Bhoyate S, Ionescu M, Kahol PK, Gupta RK (2018) Sustainable flame-retardant polyurethanes using renewable resources. Ind Crop Prod 123:480–488. https://doi.org/10.1016/j.indcrop.2018.07.025
Chen Y, Luo Y, Guo X, Chen L, Xu T, Jia D (2019) Structure and flame-retardant actions of rigid polyurethane foams with expandable graphite. Polymers 11:686–698. https://doi.org/10.3390/polym11040686
Lin Z, Zhao Q, Fan R, Yuan X, Tian F (2020) Flame retardancy and thermal properties of rigid polyurethane foam conjugated with a phosphorus–nitrogen halogen-free intumescent flame retardant. J Fire Sci 38:235–252. https://doi.org/10.1177/0734904119890685
Yang C, Shao S (2021) Rigid polyurethane foams containing modified ammonium polyphosphate having outstanding charring ability and increased flame retardancy. Front Mater 8:712809–712818. https://doi.org/10.3389/fmats.2021.712809
Agrawal A, Kaur R, Walia RS (2019) Investigation on flammability of rigid polyurethane foam-mineral fillers composite. Fire Mater 43:917–927. https://doi.org/10.1002/fam.2751
Akdogan E, Erdem M, Ureyen ME, Kaya M (2019) Rigid polyurethane foams with halogen-free flame retardants: thermal insulation, mechanical, and flame retardant properties. J Appl Polym Sci 137:47611–47624. https://doi.org/10.1002/app.47611
Howell BA, Ostrander EA (2019) Flame-retardant compounds for polymeric materials from an abundantly available, renewable biosource, castor oil. Fire Mater 44:242–249. https://doi.org/10.1002/fam.2796
Xu Z, Tao R, Mao L, Zhan J, Xiao J, Yu T (2022) Combustion and thermal properties of flame retardant polyurethane foam with ammonium polyphosphate synergized by phosphomolybdic acid. Front Mater 9:944368–944377. https://doi.org/10.3389/fmats.2022.944368
Xi W, Qian L, Li L (2019) Flame retardant behavior of ternary synergistic systems in rigid polyurethane foams. Polymers 11:207–217. https://doi.org/10.3389/fmats.2022.944368
Bo G, Xu X, Tian X, Wu J, He X, Xu L, Yan Y (2021) Synthesis and characterization of flame-retardant rigid polyurethane foams derived from gutter oil biodiesel. Eur Polym J 147:110329–110339. https://doi.org/10.1016/j.eurpolymj.2021.110329
Zagozdzon I, Parcheta P, Datta J (2021) Novel cast polyurethanes obtained by using reactive phosphorus-containing polyol: synthesis, thermal analysis and combustion behaviors. Materials 14:2699–2716. https://doi.org/10.3390/ma14112699
Acknowledgements
This work was financially the New Materials Engineering Center of Jiangsu Province Research Council of China.
Author information
Authors and Affiliations
Contributions
Junrui Chi: Writing-Original Draft, Conceptualization, Investigation. Yu Zhang: Data Curation, Visualization, and formal analysis. Fanbin Tu: Validation. Junchen Sun: Conceptualization, Data Curation. Huizhen Zhi: Writing - Review & Editing, Supervision, Funding acquisition. Jinfei Yang: Resources, Project administration.
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chi, J., Zhang, Y., Tu, F. et al. The synergistic flame-retardant behaviors of soybean oil phosphate-based polyols and modified ammonium polyphosphate in polyurethane foam. J Polym Res 30, 84 (2023). https://doi.org/10.1007/s10965-023-03447-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-023-03447-6