
Abstract
Nanoporous polymer networks of N − vinylpyrrolidone (VP) with dimethacrylates of ethylene, 1,6 − hexanediol and triethylene glycol were obtained by three-dimensional free-radical copolymerization in bulk using branched copolymers as pores templates. The branched copolymers consisted of monomers of the same type to provide their thermodynamically compatibility and solubility in the appropriate mixtures of VP − dimethacrylate. They were synthesized by radical copolymerization in toluene under chain transfer condition and characterized by the FTIR, 1H NMR, GPC, DSC and DLS methods. After curing of the stable visually homogeneous monomer-polymer mixtures, polymer composites with optical properties differed from conventional network copolymers were obtained due to the micro- or macrophase separation. According to the gravimetry, FTIR, GPC and DSC data, the macromolecular additives were partly extracted by isopropyl alcohol as a “good” solvent. The hierarchical nanoporous structure of the obtained gels was demonstrated by the SEM and low-temperature nitrogen adsorption methods. Polymer networks crosslinked with dimethacrylates of ethylene and triethylene glycol contained mostly mesopores, while pores less than 20 nm predominated in the N − vinylpyrrolidone copolymer crosslinked with 1,6 − hexanediol dimethacrylate. It was shown that the values of the specific surface area increased by an order in comparison with conventional polymer networks and reached about 11–16 m2/g.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The rapid development of innovations in biomedicine and biotechnology requires creation of biocompatible polymers of various structures including nanoporous polymer networks. They can be used as encapsulation agents for controlled drug release [1,2,3], supports for biomolecular immobilization and cell scaffolds [4,5,6,7,8,9,10], biosensors [11,12,13,14,15], etc. Among various vinyl monomers, N-vinylpyrrolidone (VP) attracts special attention as a raw material for creating biocompatible polymers for biomedical purposes [16,17,18,19], since they could be considered in a certain sense as models of natural macromolecules with amide groups. A new step in their macromolecular design is associated with the use of the template methodology for the elaboration a simple procedure for preparing the hierarchical nanoporous polymer networks based on N-vinylpyrrolidone [20].
Using this methodology, a variety of porous polymers with different nanostructures and functionalities obtained by conventional radical polymerization of various monomers either on the surface of nanostructured templates, or in the internal pores of templates has already been successfully produced [21, 22]. Typically, silica nanomaterials (e.g., silica nanoparticle and mesoporous silica), aluminum oxide, as well some polymer nanomaterials, for example, hyperbranched polymers are used as templates [23,24,25]. Despite irregularity and structure imperfection of macromolecules, they may possess the useful properties (high solubility, low solution viscosity, and high density of functional groups at the periphery of the macromolecule) similar to those of dendrimers. And, at the same time, one of their advantages is a more economic and facile synthesis [26]. In particular, they can be synthesized by polycondensation [27], ring-opening polymerization [28], living radical polymerization [29, 30], “click” reactions [31], and copolymerization of commercially mono- and divinyl monomers in the presence of a catalytic chain transfer agent [32] or a chain transfer agent (the “Strathclyde methodology”) [33]. The latter methodology allows synthesizing branched polymers with a broad variety of architectures, sizes, and functionality in a simple one-pot process [33,34,35,36,37,38,39,40,41]. Thus, copolymers were successfully prepared using branching agents of different molecular structure and functionality [34, 35], conventional chain transfer agents of various structures [33, 36]. They were obtained by copolymerization in homogeneous solution [33], emulsion [36], and suspension [37, 38]. It is possible to synthesize branched copolymers based on methacrylates and acrylamide which are sensitive to temperature and pH changes owing to wide possibilities of macromolecular design [39,40,41].
In our case [20], the templating methodology includes three-dimensional radical copolymerization of VP and triethylene glycol dimethacrylate in the presence of the purposefully synthesized copolymers with well-defined branched structure, which are compatible with the comonomers. After copolymerization the branched copolymers were removed by extraction with a solvent. The polymer framework was robust enough to withstand the high interfacial energy and strong capillary actions that could lead to pore deformation or even pore collapse after the template removing.
The purpose of the present work is to study the effect of the structure and polarity of dimethacrylate as a crosslinking agent on parameters of the porous structure of the copolymer networks of N-vinylpyrrolidone with various dimethacrylates using branched copolymers of the identical chemical nature as pore templates. Bifunctional monomers − ethylene glycol dimethacrylate (EGDM), 1,6 − hexanediol dimethacrylate (HDDM), and triethylene glycol (TEGDM) dimethacrylates, differed in length and polarity of the spacers between methacrylic groups, were used as branching and crosslinking agents in the synthesis of the soluble and crosslinked copolymers of VP, respectively. The soluble copolymers branched with the corresponding dimethacrylates were added in the VP − dimethacrylate mixture to achieve their compatibility and to control the micro- and macrophase separation under copolymerization, since both the branched copolymers, and the formed network copolymers were similar in the chemical composition.
Materials and methods
N-vinylpyrrolidone (“Alfa Aesar”) was distilled in vacuum to remove inhibitor NaOH. EGDM, HDDM, TEGDM (“Aldrich”), and 1-decanethiol (DT, “Alfa Aesar”) as a chain-transfer agent were used as received.
Soluble copolymers of VP − EGDM (br − VP − EGDM), VP − HDDM (br − VP − HDDM), and VP − TEGDM (br − VP − TEGDM) were prepared via crosslinking radical copolymerization at molar ratio of [VP]:[dimethacrylate]:[DT] = 100:12:12 in toluene as described in [42]. Their amorphous powders were isolated from the reaction mixture by precipitation with a 10-fold excess of hexane and dried in vacuum to a constant weight. The prepared copolymers (20 wt%) were dissolved, respectively, in the comonomer mixtures of VP − EGDM, VP − HDDM, VP − TEGDM of the same composition (0.6:0.4 mol fractions).
The obtained monomer-polymer mixtures were placed in glass ampoules (3 mm in diameter), evacuated, and sealed. Three-dimensional radical polymerization in bulk initiated by AIBN (0.2 wt%) was carried out at 60 °C in the absence and in the presence of the soluble copolymer additives. To prevent spontaneous copolymerization, a stable TEMPO radical ((3–4)∙10−4 M) was added to the reaction mixture. As a result, in the absence and in the presence of copolymer additives, respectively, the VP − dimethacrylate network copolymers (nc − VP − dimethacrylate) and polymer composites (pc − VP − dimethacrylate) were obtained.
Characterization
FTIR and 1H NMR spectroscopy
The compositions of br − VP − EGDM, br − VP − HDDM, and br − VP − TEGDM were studied by IR-spectroscopy using a Bruker α FTIR equipment, and by 1H NMR using an AVANCE III 500 MHz Bruker BioSpin superconducting pulsed broadband two-channel NMR spectrometer. IR spectral analysis was performed using films of the copolymers casting from chloroform solutions on KBr glasses. For 1H NMR studies, the copolymer solutions in deuterated chloroform (6 mg/ml) were prepared.
GPC method with two detectors
The molecular weights of the branched copolymers and the sols isolated from the copolymer composites were measured by GPC as described in [42] with the use of a Waters GPCV 2000 chromatograph (two PL-gel columns, 5 μm, MIXED-C, 300∙7.5 mm) equipped with two detectors − a refractometric detector with Empower Pro software and a light scattering detector with Astra software.
DSC method
The glass-transition temperatures Tg of the branched copolymers and the gels isolated from the copolymer composites were measured by the DSC method using a METTLER TOLEDO STARe instrument with DSC 822 modulus at a heating rate of 5 °C/min.
DLS method
The dynamic light scattering method was used to study the solutions of the branched copolymers of various concentrations in isopropyl alcohol, as well as the monomer-copolymer mixtures containing 1 wt% of the branched copolymers at a detection angle of 90°. A Photocor Compact (Photocor Instruments Inc., USA) equipped with a diode laser, operating at a wavelength of 654 nm, was employed. The solutions of the branched copolymers in isopropyl alcohol were previously passed through a filter with pores of 0.45 μm in diameter. Before measuring, the vial with the solution was thermostated for ca. 20 min. The autocorrelation functions of the scattered light determined in the dynamic light scattering regime were processed using "DynaLS" software, which allowed analyzing distribution functions of the particles in solution depending on correlation times tc and diffusion coefficients D. The hydrodynamic radii Rh of the branched copolymers in isopropyl alcohol were calculated using the Einstein-Stokes equation
where D is the diffusion coefficient, k is the Boltzmann constant, T is the absolute temperature, and η is the viscosity of the medium, in which the dispersed particles are suspended. The dependences of the light scattering intensity distribution versus the correlation times tc, the diffusion coefficients D, and the hydrodynamic radii Rh were plotted.
The isothermal calorimetry
The kinetics of copolymerization of VP − dimethacrylate in the absence and in the presence of the branched copolymers was investigated by isothermal calorimetry using a DAK-1-1 instrument. The values of the double bonds conversion and reduced copolymerization rate w/[M] were calculated according to the procedure described in [43], taking into account the molar heats of VP and MMA polymerization in bulk equal to Q1 = 64.3 kJ/mol and Q2 = 58.8 kJ/mol, respectively. The Q1,2 values for copolymerization were calculated by the additive scheme:
where n1, n2 are the molar fractions of VP and dimethacrylate in the initial mixture.
When the values of the copolymerization heat were averaged according to the additive scheme, a certain error, increasing with the difference in the molar heats of the monomers, was included. The rate was reduced to the current averaged concentration of the monomer mixture.
Scanning electron microscopy
The surface of the network copolymers after extraction of polymer additives was studied by SEM on a Zeiss Leo Supra 25 instrument. For this purpose, the end surface of cylindrical samples was sprayed with ultrafine carbon.
The sol−gel analysis
The sol−gel analysis of pc − VP − EGDM and pc − VP − HDDM was carried out in the Soxhlet extractor for 14 h, and of pc − VP − TEGDM − for 7 h at the boiling point of isopropyl alcohol as a good solvent for the branched copolymers. The isolated soluble (sols) and insoluble (gels) products were dried in vacuum to a constant weight. The compositions of the gels and the sols were studied by the FTIR method using a "Bruker α" equipment in the transmission mode. The sols were placed on the glasses and evacuated from the solvent; the gels were prepared as suspensions in vaseline oil.
The BET analysis
After sol extraction, the porous structure of the previously evacuated at ca. 100 °C gels was investigated by low-temperature nitrogen adsorption on an Autosorb-1 analyzer (Quantachome Instruments corp., USA). The specific surface area (Ssp) of the gels is determined using the Brunauer-Emmett-Teller (BET) equation:
where P is the nitrogen pressure in the sample cell, P0 is the saturated nitrogen vapor pressure at 77 K, W is the weight of nitrogen absorbed at a given value of P/P0, Wm is the weight of adsorbate in the surface monolayer, C is the parameter of the BET equation characterizing the adsorbent-adsorbate interaction. The value of total pore volume Vp was determined by measuring the amount of nitrogen adsorbed at the value of P/P0 close to 1. The pore size distribution curves were plotted using the Barrett, Joyner, and Halenda (BJH) method [44]. The error in Vp and Ssp values determination did not exceed 7%.
Results and discussion
Synthesis of the branched copolymers, their structure and physicochemical properties
The branched copolymers were prepared by radical copolymerization of VP with various dimethacrylates in toluene in the presence of a chain transfer agent – DT (Scheme 1). The reaction proceeds with a high yield (Table 1). The obtained copolymers were soluble in polar and low polar solvents (water, alcohols, chloroform, etc.).

The synthesis of the copolymers of VP − dimethacrylates by radical copolymerization in toluene in the presence of DT
According to IR-spectroscopy data on the kinetics of the separate conversion of the components of the reaction mixture, EGDM was more reactive in radical copolymerization than VP [43]. These conclusions are more reliable than those based on the values of copolymerization constants defined for methyl methacrylate as a monofunctional analogue. The effective constant of copolymerization of “pendant” C=C bonds of dimethacrylates can change significantly in crosslinking copolymerization.
As a result, in the initial stages of copolymerization all radicals attach dimethacrylates and polymer chains are formed, consisting mainly of dimethacrylate units. Attaching polymer radicals to pendant double bonds of dimethacrylates leads to the appearance of side branches in the polymer chain and to the formation of branched structures (Scheme 1). In the presence of DT, the primary polymer radicals become shorter due to the chain transfer reaction, and the probability of the macrogel formation decreases. According to elemental analysis, the prepared copolymers contained sulfur atoms (Table 1). The experimental sulfur content was less than the calculated value because of some amount of −SC10H21 groups contained in the low-molecular fraction (soluble in hexane) enriched with dimethacrylates. Furthermore, DT was consumed incompletely in the chain transfer reaction [42].
With the increase of the conversion degree, after depletion of dimethacrylate in the mixture of the monomers, the polymer chains appear, consisting mainly of VP-units. In addition, at deep copolymerization stage one can expect the appearance of VP polymer. As a result, compositionally heterogeneous polymer products were formed; their macromolecules differed markedly by solubility in toluene. Thus, after the synthesis completion, br − VP − EGDM was entirely soluble in toluene, and the reaction mixture was visually clear. At the same time, br − VP − HDDM and br − VP − TEGDM had limited solubility in toluene and, as a result, the reaction mixtures were opalescent. Moreover, a toluene-insoluble fraction of br − VP − TEGDM was isolated from the reaction mixture.
Some conclusions on the structure of the prepared copolymers were made by analyzing their IR spectra (Fig. 1S, Supplementary Materials). The specific absorption bands of C=O bonds in the methacrylate group of br − VP − EGDM, br − VP − HDDM, and br − VP − TEGDM were observed at ν ~1728, 1723, and 1726 cm−1, respectively. The absorption band of C=O bonds in the lactam cycle of VP − units for all copolymers was observed at ~1680 cm−1. To evaluate the composition of the studied copolymers, the optical densities at the maximum of the absorption bands were measured, and their ratios (A) were calculated (Table 1). It can be concluded that the prepared copolymers were similar in composition. This is also confirmed by elemental analysis and the calculated monomer composition (Table 1).
The copolymers were also investigated by 1H NMR spectroscopy (Fig. 2S, Supplementary Materials). As expected, signals related to monomer units were observed in the spectra. Thus, there were two groups of bands related to VP units; the first set included signals due to NCHα protons in the polymer chain and –CH2C=O fragments of pyrrolidone at δ = 3.0–4.0 ppm. The second one consisted of signals attributed to protons of CH2 in the polymer chain, as well as to C–CH2–C and NCH2 fragments of pyrrolidone at δ = 1–3 ppm. Signals of CH3-group in dimethacrylate units were detected in the spectrum of the copolymers at δ~1 ppm. Signals at δ = 1.3 and 2.5 ppm may correspond to protons of (CH2)8 and CH2–S fragments of DT. Signals of hydrogen atoms in –CH2–CH2 fragment of dimethacrylate involved in reaction were observed in 1H NMR spectra at δ~4.1 ppm. Weak signals detected at 5.7 and 6.2 ppm were corresponded to vinyl hydrogens on pendant unreacted C=C bonds in dimethacrylates (Fig. 2S, inset, Supplementary Materials) [35]. In accordance with ozonolysis data [42], the similar object contained ca. 0.56∙10−3 mol of double bonds per gram of the copolymer or about 11 mol C=C bonds per mole of the copolymer with Mw ca. 20∙103. These can be either pendant unreacted C=C bonds of dimethacrylates, or C=C bonds resulting from the polymer chains termination by disproportionation.
Figure 1a shows molecular weight distribution (MWD) curves of the soluble copolymers of VP − dimethacrylate. For comparison, the curve for linear PVP is given. It can be seen that the MWD curves of the copolymers are multimodal and consist of several overlapping peaks at low molecular weight area, which indicates their high degree of molecular weight heterogeneity. The average molecular weight Mw and molecular weight at the peak maximum Mp (a significant parameter for compositionally heterogeneous copolymers), calculated from the data of two detector method, are given in Table 2. The absolute values of Mw and Mp for br − VP − HDDM are higher than those for br − VP − EGDM and br − VP − TEGDM. The growth of br − VP − HDDM polymer chains probably proceeds under conditions favorable for increasing their length and molecular weight.

Molecular weight distribution curves of the soluble VP − dimethacrylate copolymers and PVP (a); the dependences of their molecular weights Mw on elution volume VR in semilogarithmic coordinates (b)
Some conclusions about the topology of the studied copolymers were made from the analysis of the semilogarithmic dependences of Mw on the elution volume VR (Fig. 1b). The curves can be divided into several regions. In the first region with small values of VR~5.5–6.5 ml, macromolecules with molecular weight above ~105–106 are eluted. In this region, at the same VR value, a part of macromolecules of br − VP − EGDM and br − VP − HDDM with a lower molecular weight than that of linear PVP is eluted. They probably represent nanogel particles with a high content of dimethacrylates – internally crosslinked and slightly swelling in the eluent. It can be seen that the part of macromolecules of br − VP − TEGDM with Mw~106 has a dense packing of the chains due to the branched structure. At the same VR value, they have a higher molecular weight than linear PVP. In the second region, in the range of VR~6.5–7.7 ml, the Mw(VR) curves for br − VP − EGDM and for linear PVP practically coincide, indicating their similar structure. The curves for br − VP − HDDM and br − VP − TEGDM lie higher than that for linear PVP, and the topology of their macromolecules is likely to be branched. At VR values above 7.7 ml, the copolymers contain low-molecular weight macromolecules of similar topology.
The factor g’ is defined as the ratio of the root-mean-square gyration radius (rms) of the copolymer to that of linear PVP and characterizes its branching. The value of g’ for macromolecules with molecular weight less than 3∙104 lies in the interval of 0.5–0.64, while for the linear polymer g’ = 1. The small values of rms~10 nm for br − VP − EGDM and br − VP − HDDM with Mw more than 3∙104 indicate their close packing caused by a branched nature. Thus, the studied copolymers differ in molecular weight and topology; they consist of macromolecules with various branching degree and nanogel particles.
Important information on the copolymers studied was obtained from the analysis of their thermograms and the values of glass transition temperature Tg, which were closely related to the molecular weight, the topology of macromolecules, and the water content. Figure 3S, Supplementary Materials shows DSC curves for the first and the third scanning of the studied copolymers. In the region of ca. 40–140 °C, for the first scan of all copolymers the DSC curves have a wide endothermic peak, apparently associated with the loss of water. According to IR-spectra in the region of 3400–3600 см−1 (Fig. 1S, Supplementary Materials), it forms hydrogen bonds to the copolymers. It can be seen that this peak overlaps with the step of the change in the heat capacity due to the phase transition, namely, the glass transition.
According to the literature data [45], PVP is considered to be dry after the first scanning of the sample to 200 °C. In general, this assumption is also fulfilled for all studied copolymers. So, the glass transition temperatures of the copolymers were determined during the second and the third scanning and were fairly similar. Here, we present the result of the 3-th scan as more reliable, because no peaks associated with water evaporation were observed on the thermograms. The steps on the thermograms were considered as second-order phase transitions, and the glass transition temperatures of copolymers, Tg, were determined by a DIN standard [46] (Table 2). The second step on the thermograms is weak, and the Tg2 value is less reliable (especially for the br − VP − TEGDM copolymer), so we do not show it in Table 2. As will be shown below, the experimental Tg values will allow to identify these copolymers in the gels isolated from polymer composites.
All copolymers have an α-transition in the low-temperature region due to the defrosting of the mobility of polymer chains with branches. Moreover, in the high-temperature region, there is the second α-transition, which can be caused by the defrosting of molecular mobility in linear PVP. Here, we estimated the range of Tg values as 125–150 °C for linear PVP with Mn ca. (9–13)∙103. Therefore, it can be assumed that the studied copolymers contain a linear PVP formed during the copolymerization. However, the second glass transition temperature can also be interpreted as the α-transition in nanogel particles, in which the mobility of polymer chains is frozen due to intramolecular crosslinking or their strong physical interactions.
The studied copolymers consist of high polar (VP) and low polar (dimethacrylates) monomer units; in addition, there are nonpolar –SC10H21 groups at the ends of the polymer chains terminated by the chain transfer mechanism. The difference in polarity of these units determines the amphiphilic character of the copolymers and their ability to self-organization with the formation of polymer aggregates in polar media. It is known [47] that the mechanism of aggregation of amphiphilic macromolecules may differ, depending on the composition, the diphilicity, and the topology of macromolecules. Thus, the amphiphilic macromolecules of branched topology exist in polar solvent as monomolecular micelles and their aggregates (multimolecular micelles), or they can undergo intra-phase separation and form stable micelle-like aggregates similar to the traditional block copolymers. The investigated compositionally heterogeneous copolymers of VP − dimethacrylates of different topology seem to exist in polar media as monomolecular micelles, multimolecular micelles, as well as micelle-like aggregates. They have a core formed by the fragments of polymer chains enriched with dimethacrylate units. The micellar nature of macromolecules of some branched copolymers in water was demonstrated in work [48], where fullerene C60 and its small clusters were found in a hydrophobic core of the polymer particles by the cryo-TEM method.
At first, we studied the behavior of the branched copolymers in polar VP and in isopropyl alcohol as a model polar solvent by the DLS method. Polar VP was a good solvent for the copolymers, and their diluted solutions containing separated macromolecules dispersed light poorly. However, they were self-organized in isopropyl alcohol, what evidenced by the dependences of the average intensity of light scattering I on the copolymer concentration c (Fig. 4S a, Supplementary Materials). In the studied concentration range, I(c) curves of the investigated copolymers (Fig. 4S a, Supplementary Materials) have one or two break points, which should be interpreted as critical concentrations of aggregation. In the solutions of br − VP − EGDM and br − VP − HDDM at concentrations of ~0.3 and ~0.4 wt%, respectively, the value of I increases, evidently, due to the aggregation growth. For br − VP − TEGDM, there are two breaks at the I(c) dependence at concentrations of ~0.1 and ~0.6 wt%, indicating a more complex solution structure due to the strong compositional heterogeneity of its macromolecules. Low polar macromolecules with a high TEGDM content seem to be aggregated at low concentrations, whereas more polar macromolecules are aggregated at high concentrations of this copolymer.
The values of the hydrodynamic radii Rh of scattering centers in 1% solutions of the studied copolymers in isopropyl alcohol are given in Table 2. The solutions have nano- and micron-sized scattering centers (individual macromolecules and their aggregates), the contribution of the latter to light scattering reaches ca. 60% in the solutions of br − VP − EGDM and br − VP − HDDM. In the solution of br − VP − TEGDM, light scattering occurs mainly due to the scattering centers of small size (70%). Thus, br − VP − EGDM and br − VP − HDDM are more prone to aggregation in isopropyl alcohol than br − VP − TEGDM. This can be explained by a high diphilicity of their macromolecules containing low-polar units of EGDM and HDDM, and, therefore, by declining of their thermodynamic affinity with the solvent.
Figure 4S b, Supplementary Materials shows I(c) dependences for the mixtures of VP − dimethacrylate containing the corresponding branched copolymers. The experimental points on these dependencies can be satisfactorily approximated by only one linear function. The corresponding monomer mixtures appear to be thermodynamically good solvents for these copolymers, and only individual macromolecules are present in the monomer mixtures. However, the experimental points on the I(c) dependence for the mixture of VP − HDDM with br − VP − HDDM additive lies much higher, and their considerable dispersion is caused probably by changes in the concentration of aggregates during filtration of the solution.
The dependences of the light intensity distribution I on the correlation times tc and on the diffusion coefficients D for the investigated mixtures differ significantly (Fig. 2). The distribution curves of I(tc) and I(D) for the mixture of VP − HDDM exhibit narrow peaks with the maxima at ~18 ms and 13∙10−10 cm2/s, respectively. Such small values of diffusion coefficients are usually characteristic for light scattering with Rh ~103 nm. Consequently, in the VP − HDDM monomer mixture there are highly structured regions formed by HDDM molecules in the polar monomer. The distribution curves of I(tc) and I(D) for the mixture of VP − HDDM with br − VP − HDDM additive have also narrow peak with the maxima at ca. 26 ms and 9∙10−10 cm2/s, respectively. The polymer additive leads to an expected reduction in the value of D by a factor of 1.4 and an increase in the tc value due to increasing the medium viscosity. At the same time, the associative structure of the VP–HDDM monomer mixture is not destroyed.

Distribution of the light scattering intensity I by correlation times tc (a) and diffusion coefficients D (b) for the monomer − copolymer mixtures at 20 °C. The content of the branched copolymer additive is 1 wt%
Thus, the VP − dimethacrylate copolymers were soluble in polar media. They contained branches in side chains and were characterized by the same monomer composition, similar molecular weight, and more than one glass transition temperature due to the compositional heterogeneity of the macromolecules. In dilute solutions of isopropyl alcohol, amphiphilic copolymers were represented as individual macromolecules (monomolecular micelles) with hydrodynamic radii ca. 3–5 nm and multimolecular micelles; copolymers with low-polar EGDM and HDDM units were more aggregated. Aggregation of the VP − EGDM and VP − TEGDM copolymers was suppressed in the corresponding monomer mixtures. In turn, the mixture of VP − HDDM with br − VP − HDDM additive was a highly ordered system contained light scattering centers of micron size. Individual macromolecules and their aggregates, apparently, can serve as pores templates. So, the next step was to prepare polymer networks and to determine parameters of their porous structure.
Formation of the porous polymer networks
Although all monomer-copolymer mixtures are visually transparent, they represent colloid systems, which are stable in time and not destroying into separate phases due to the high thermodynamic compatibility of polymer additives consisting of the same monomers as the VP − dimethacrylates mixtures.
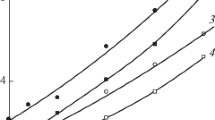
Figure 3 shows kinetic curves of polymerization of the VP − dimethacrylate mixtures with and without the polymer additives. All monomer mixtures are copolymerized in autoacceleration and autodeceleration modes caused by freezing of the mobility of the reagents and pendant C=C bonds. A comparison of the curves for the copolymer mixtures indicates that the dimethacrylate structure effects the process rate, the beginning of structure formation, and the limiting conversion of C=C bonds. A high mobility of the monomers and pendant C=C bonds is maintained in the course of copolymerization of VP with HDDM or TEGDM as crosslinking agents. As a consequence, the maximum reduced reaction rate is achieved at the values of conversion C equal to 33 and 39%, respectively. When VP copolymerizes with EGDM, the structure formation begins earlier and the maximum reduced rate is reached at C ~11%. At the same time, the VP − HDDM system exhibits specific behavior during copolymerization: the maximum values of the reduced rate and the limiting conversion of C=C bonds (85%) are observed. It should be noted that during homopolymerization these values increase in the line of EGDM<HDDM<TEGDM, as expected, according to their length, flexibility, and effective reactivity. This effect is, apparently, due to such peculiarity of VP − HDDM mixture as a highly ordered system. In the polar monomer HDDM exists in the form of associates, what is in a good agreement with the DLS data (Fig. 2). Such HDDM associates can act as nano- and microreactors, in which heterogeneous copolymerization proceeds at higher rates than in more homogeneous VP − EGDM and VP − TEGDM mixtures.

Dependences of the reduced rate of copolymerization w/[M] on the conversion C in the absence and in the presence of the copolymer additives at 60 °С. Concentrations of AIBN and TEMPO are 0.2 wt% and 4∙10−4 M, respectively
The character of copolymerization is generally maintained in the presence of the corresponding polymer additives. However, the reduced rates are significantly decrease, the w/[M](C) curves shift to the area of smaller conversion values, and the maximum rates are observed at С ~ 4 and 24%, respectively. The rate decrease is apparently caused by the influence of the polymer additive on the initiation rate constant, or its inhibitory effect. Polymer additives containing pendant C=C bonds of dimethacrylate units are probably inactive in three-dimensional radical copolymerization. This is favored by micellar character of macromolecules with core formed by dimethacrylate fragments. As a result, C=C bonds of dimethacrylate concentrated in the core remain inaccessible for interaction with growing radicals. It should be noted, that the highest rate is also observed during copolymerization of the mixture of VP − HDDM with br − VP − HDDM. Branched copolymer does not destroy the structure of the monomer mixture what affects the copolymerization rate.
After copolymerization, polymer composites with the homogeneous distribution of the additives in the polymer body were obtained. According to isothermal calorimetry, the limiting conversions of double bonds in pc − VP − EGDM, pc − VP − HDDM, and pc − VP − TEGDM were ca. 90, 84 and 77%, respectively. The polymer composites represented chemical networks of various crosslink densities and free volumes with the end chains consisted of VP units. The crosslink density of chemical network is determined by the dimethacrylate structure, and it is the reciprocal value of its molecular weight. Therefore, the polymer composite based on EGDM and HDDM have a more dense cross-linked and is characterized by small free volumes compared to pc − VP − TEGDM. The appearance of PVP should be expected after the depletion of the active comonomer during three-dimensional copolymerization both in the absence, and in the presence of the polymer additives. Optical properties of the polymer monoliths occurred to be different. The samples of pc − VP − EGDM, and pc − VP − HDDM were opaque white due to a strong scattering of visible light, whereas pc − VP − TEGDM was only opalescent. These optical changes indicate the phase separation induced by three-dimensional radical copolymerization of the monomer−polymer mixtures. During pc − VP − EGDM, and pc − VP − HDDM preparation, the macrophase separation is more pronounced due to the formation of three-dimensional structures, crosslinked by dimethacrylates with short oligomeric spacers between methacrylic groups, and the polymer additives are separated into discrete regions, which refractive indexes are significantly differed from those of the network structures. It should be noted that nc − VP − EGDM, nc − VP − HDDM, and nc − VP − TEGDM obtained in the absence of polymer additives were transparent, and the microphase separation between the formed copolymer networks and soluble polymer products, for example PVP, was insignificant. These processes determine the resultant morphology and allow us to control the monolith structure [49].
Sol−gel analysis of the polymer composites
The copolymer composites were extracted with isopropyl alcohol as a "good" solvent for branched copolymers. After extraction, all samples were opaque white monoliths without changing their shape and size. The monolith did not mechanically destroyed at high temperatures, but some structural changes were observed caused by limited swelling. In all cases, the sol content was two times higher than expected (20 wt%) under the assumption that the polymer additives did not participate in copolymerization.
The IR spectra of both the sols separated from various polymer composites, and the initial polymer additives were recorded (Fig. 4). The IR spectra of br − VP − EGDM and the sol separated from pc − VP − EGDM were similar (Fig. 4a), the individual characteristic bands of stretching vibrations of C=O groups in EGDM and VP units were observed. It is obvious that br − VP − EGDM was present in the analyzed sol. However, the IR spectra of the sols separated from the pc − VP − TEGDM and related branched copolymers differed noticeably (Fig. 4b). In the IR spectra of the sol, the band of stretching vibrations of C=O bond in TEGDM units was represented as a shoulder on the C=O absorption band of the lactam cycle at ca. 1660 cm−1. A shift in the frequency of stretching vibrations of C=O bond in VP units to lower frequencies in comparison with the initial copolymer additives, as well as the broad absorption band of the stretching vibrations of OH group with a maximum at 3300 cm−1 were also observed. Compared to br − VP − TEGDM, the maximum of the absorption band of OH groups is shifted by 150 cm−1. In addition, there are some changes in the IR spectrum of the sol in the range of 1200–1100 cm−1. These may be caused by a strong hydrogen bonding of methacrylic C=O groups with isopropyl alcohol and adsorbed water, or even by a chemical transformation of -C-O-C bonds in TEGDM units during copolymer extraction. According to elemental analysis, the nitrogen content was 2.6%, and the sol was different from br − VP − TEGDM in the monomer composition. The 1H NMR spectrum of the sol confirms the low content of the VP units in the polymer chains and the residues of the DT, judging by the signals of the H-protons of –CH2-S- groups at 2.5 ppm. It is very likely, that formed PVP is also extracted from the polymer networks. This leads to a further change in the sol composition.

IR spectra of branched copolymers (a) and sols extracted from various polymer composites (b)
The assumption of the presence of a new polymeric product in the sols is confirmed by GPC data obtained from refractometer detector, what allows us to determine products with low molecular weights. Figure 5 shows chromatograms of the branched copolymers and the sols extracted from the related polymer composites. In addition to a product with high molecular weight, there is a low-molecular oligomer with Mp ca. 500 or 700. It is probably a product of VP homopolymerization after depletion of active dimethacrylate in radical copolymerization. Thus, the molecular weight distribution of the extracting polymer additives varies. As can be assumed from the IR-spectroscopy and GPC data, the sols separated from polymer composites contain a high-molecular weight fraction of the copolymer additives, PVP, and the residual solvent bound by H-bond to C=O groups of the soluble copolymer. We found that template was washing out during pc − VP − TEGDM extraction with water. This was confirmed by a decrease in the sample mass, as well as by the appearance of the intense C=O absorption bands of TEGDM units in the IR spectrum.

Chromatograms of the branched copolymers and the sols extracted from the polymer composites by isopropyl alcohol
The content of the gels after pc − VP − EGDM and pc − VP − HDDM extraction exceeded the theoretically possible value, i.e. 80%, if all monomers participated in the network formation. They appeared to contain the solvent and partially retained the branched copolymer additives. The content of the gel after pc − VP − TEGDM extraction was below than expected (58%). Apparently, during copolymerization of VP and TEGDM in the presence of br − VP − TEGDM, soluble PVP was formed in a larger amount than in the other two systems.
Some conclusions on the gel composition were made from the analysis of their IR-spectra (Fig. 5S, Supplementary Materials) and of A values. The gels after pc − EGDM−VP, pc − HDDM−VP, and pc − TEGDM−VP extraction were characterized by A values of 0.9, 1.21 and 1.33, respectively. Perhaps, a residual template with a high content of dimethacrylate units contributed to the VP − HDDM and VP − TEGDM gel composition. Judging from a wide absorption band in the range of 3600–3000 cm−1 in the IR-spectra, adsorbed water and isopropyl alcohol form hydrogen bonds with C=O groups in VP units of different strength and energy. As a consequence, the vibration frequencies of C=O bonds for the studied gels were 1660, 1678, and 1670 cm−1, respectively. Thus, the gel after pc − VP − EGDM extraction with the low conversion of C=C bonds is the most favorable for the diffusion of polar molecules (alcohol, absorbed water), which are firmly bound by hydrogen bond with C=O groups of the copolymer.
Taking into consideration the IR data of the studied gels, a three-fold DSC scanning of the samples in the temperature range of 20–180 °C was carried out to remove low molecular components (residual solvent and absorbed water), as well as to determine their glass transition temperature and to verify their compositional heterogeneity. During the first scan, wide endothermic peaks associated with removal of water and alcohol were observed (Fig. 6S a, Supplementary Materials). At the third scan (Fig. 6S b, Supplementary Materials) these peaks disappeared completely, and the steps observed on the thermograms were caused by glass transition in the investigated samples. In the region of 50–70°С, low-temperature transitions were observed which apparently caused by the defrosting of the mobility of the residual polymer additives. The determined glass transition temperatures of 66, 55, and 54 °C correlated well with the Тg values related to branched copolymers (Table 2). The second transition took place in the high-temperature region and was apparently associated with the defrosting of the polymer network mobility. However, it was not sufficiently intensive to reliably determine the Tg value of the polymer network. The DSC data confirm that polymer additives are not fully extracted from densely crosslinked polymer networks. It may be caused by three-dimensional radical polymerization of VP and dimethacrylates in the internal volume of branched templates; as a result, their macromolecules are included in the structure of the polymer network. Thus, it is not possible to remove completely the polymer additives under given extraction conditions: they are partly retained in polymer gels and can affect the parameters of their porous structure.
The porous structure of the gels isolated from the polymer composites
It is reasonably to assume that after removing the polymer additives from the polymer composites, pores should appear in the gels. Indeed, SEM micrographs confirm the presence of pores between polymer agglomerates (Fig. 6). The micrographs clearly show interconnected pores of various sizes permeating the polymer body. The gel of pc − VP − HDDM consists of larger agglomerates compared to the gels of pc − VP − EGDM and especially of pc − VP − TEGDM. It is apparently caused by kinetic features of copolymerization and the growth of polymer chains in the highly structured system.

Micrographs of the end surfaces of the gels isolated from pc − VP − EGDM (a), pc − VP − HDDM (b), and pc − VP − TEGDM (c) obtained by the SEM method
The stages of the preparation of VP porous networks in the presence of a polymer additive are shown in Scheme 1S, Supplementary Materials. As a result of the phase separation induced by three-dimensional radical copolymerization of VP with dimethacrylate, the polymer additives are separated into micro- and macrophases of different sizes. Owing to the swelling of the polymer network, they are extracted with a solvent and partially released leaving pores. Taking into account the size of individual macromolecules and their aggregates determined in polar solvent by DLS, it can be assumed that primarily mesopores should be present in the prepared gels.
The quantitative characteristics of the porous structure of the prepared gels, such as pore size, total pore volume Vp, and specific surface area Ssp, were estimated by the low-temperature nitrogen adsorption method. Figure 7 shows the adsorption and desorption isotherms of nitrogen after evacuation of volatile components from the gels. They can be attributed to type IV [44]. The obtained isotherms practically coincide, which indicates the close porosity of the studied gels. The hysteresis on the curves indicates their mesoporous structure.

Adsorption-desorption isotherms of the prepared gels
As it follows from Table 3, the Ssp value increases in the line of the gels separated from pc-VP-EGDM, pc-VP-HDDM, and pc-VP-TEGDM. This can be due to the formation of large amount of the sol, the removal of which leads to increase in the specific surface area of the resulting polymer network VP − TEGDM. However, such correlation is not observed for the case of the pore volume. It is possibly that the pores of a small size are unstable when the samples are drying from the solvent.
As follows from the curves of the pore size distribution (Fig. 8), nanopores appear in the prepared gels. All the curves have two peaks indicating the presence of pores of different size including micropores (up to 2 nm in diameter), small mesopores or submicropores (from 2 to 4 nm), mesopores (up to 50 nm), and macropores (over 50 nm) [21]. Micropores appear as a result of the polymer chains crosslinking in the polymer framework; they can also be formed due to internal stresses and shrinkage in the polymer networks during the three-dimensional copolymerization and the solvent removal; the walls of the pores converge, and even their complete closure is possible. The appearance of the mesopores in the gels is obviously caused by the release of the dispersion phase of various sizes formed by both the individual macromolecules, and their aggregates. Macropores are formed due to the appearance of large cavities occupied by the solvent and its subsequent removal. The meso and macropores compose the transport porous structure of the obtained polymer networks. Thus, the VP polymer networks, prepared using various dimethacrylates as crosslinking agents, represent hierarchical nanoporous structures, which simultaneously possess micro-, meso-, and macropores. The pore size distribution curves in the gels isolated from pc − VP − EGDM and pc − VP − TEGDM practically coincide. At the same time, mesopores less than 20 nm predominate in the gel isolated from pc − VP − HDDM. Evidently, HDDM as a crosslinking agent is useful for preparing a polymer network with small pores. Possibly, this is caused by specific conditions for preparation of this copolymer composite when copolymerization of VP with HDDM takes place in micro- and nanoreactors formed by a low-polar monomer in polar VP. This leads to an increasing the size of the polymer particles presented on the micrographs of this gel (Fig. 6c) and to decreasing the pore size. The VP − TEGDM network copolymers obtained in the absence of the pore templates have practically no porous structure; their Ssp value is less than 1 m2/g. Thus, in this study polymer monoliths of VP with dimethacrylates of various structure and polarity with a noticeable specific surface and a hierarchical porous structure were prepared.

Pore size distribution curves of the gels isolated from the polymer composites
Conclusions
The monolithic polymer networks of VP − dimethacrylates with controlled hierarchical nanoporous structure have been prepared by three-dimensional radical copolymerization using branched copolymers of VP − dimethacrylates as pore templates. These branched copolymers are thermodynamic compatible and high soluble in the monomer mixtures. After copolymerization of the monomer-polymer mixtures, polymer composites with the homogeneous distribution of the additives in the polymer body and high conversion of the C=C bonds were obtained. The polymer additives were separated into discrete regions of different size and refractive index due to the micro- or macrophase separation. Then they were extracted by a “good” solvent. The gels are slightly swollen in polar media owing to the high crosslinking degree and mechanically stable. The Ssp values increase by an order in comparison with conventional polymer networks formed in the absence of the macromolecular additives. The resulting VP − TEGDM monolith was characterized by a maximum specific surface area and demonstrated large number of mesopores, whereas the VP − HDDM monolith obtained under copolymerization in highly structured media contained mainly small pores. Thus, we have established a new factor to control the porous structure of VP polymer networks and to increase the micropores and submicropores content using low polar HDDM as a crosslinking agent. The monoliths of VP − dimethacrylates are promising for applications in aqueous solution, and these materials could be useful for the solving of many biomedical and biotechnological problems e.g., for rapid mass transport and for controlled releasing drugs as encapsulation agents. The initial monomers are easy commercially available, and the methodology for the production of template pore agents and porous polymer monoliths is simple. In this work, we propose a route to hierarchically nanoporous polymer monoliths of VP with tunable morphology by means of templating methodology that is apparently applicable to various network polymers based on a wide range of vinyl monomers.
References
Abidian MR, Kim DH, Martin DC (2006) Conducting-polymer nanotubes for controlled drug release. Adv Mater 18:405–409
Fenton OS, Olafson KN, Pillai PS, Mitchell M, Langer R (2018) Advances in biomaterials for drug delivery. Adv Mater 30:1705328–1705356
Hossen S, Hossain MK, Basher MK, Mia MNH, Rahman MT, Uddin MJ (2018) Smart nanocarrier−based drug delivery systems for cancer therapy and toxicity studies: a review. J Adv Res 15:1–18
Kimmins SD, Cameron NR (2011) Functional porous polymers by emulsion templating: recent advances. Adv Funct Mater 21:211–225
Tripodo G, Marrubini M, Corti G, Brusotti C, Milanese M, Sorrenti L, Catenacci G, Calleri E (2018) Acrylate−based poly−high internal phase emulsions for effective enzyme immobilization and activity retention: from computationally−assisted synthesis to pharmaceutical applications. Polym Chem 9:87–97
Xiao Y, Zhou M, Mi Z, Liu W, Zhou Y, Lang M (2018) Hepatocyte culture on 3D porous scaffolds of PCL/PMCL. Colloids Surf B: Biointerfaces 173:185–193
Lee A, Langford CR, Rodriguez-Lorenzo LM, Thissen H, Cameron NR (2017) Bioceramic nanocomposite thiol−acrylate polyHIPE scaffolds for enhanced osteoblastic cell culture in 3D. Biomater Sci 5:2035–2047
Murphy AR, Ghobrial I, Jamshidi P, Laslett A, O'Brien CM, Cameron NR (2017) Tailored emulsion−templated porous polymer scaffolds for iPSC-derived human neural precursor cell culture. Polym Chem 8:6617–6627
Wang A, Paterson T, Owen R, Sherborne C, Dugan J, Li J, Claeyssens F (2016) Photocurable high internal phase emulsions (HIPEs) containing hydroxyapatite for additive manufacture of tissue engineering scaffolds with multi-scale porosity. Mater Sci Eng 67:51–58
Oh BHL, Bismarck A, Chan Park MB (2014) Injectable, interconnected, high porosity macroporous biocompatible gelatin scaffolds made by surfactant free emulsion templating. Macromol Rapid Commun 36:364–372
Zhao C, Danish E, Cameron NR, Kataky R (2007) Emulsion-templated porous materials (PolyHIPEs) for selective ion and molecular recognition and transport: applications in electrochemical sensing. J Mater Chem 17:2446–2453
Silverstein MS, Tai H, Sergienko A, Lumelsky Y, Pavlovsky S (2005) PolyHIPE: IPNs, hybrids, nanoscale porosity, silica monoliths and ICP−based sensors. Polymer 46:6682–6694
Li Z, Yang Y−W (2017) Creation and bioapplications of porous organic polymer materials. J Mater Chem B 5:9278–9290
Gendrikson OD, Zherdev AV, Dzantiev BB (2006). Russ Biol Chem Rev 46:149–192
Dmitrienko EV, Pyshnaya IA, Pyshnyi DV, Martyanov ON (2016) Molecularly imprinted polymers for biomedical and biotechnological applications. Russ Chem Rev 85:513–536
Panarin EF (2017) N-vinylamides and related polymers as delivery agents of biologically active compounds. Russ Chem Bull 64:15–23
Panarin EF (2017) Biologically active polymer nanosystems. Russ Chem Bull 66:1812–1820
Solovskii MV, Borisenko MS, Ershov AY, Zakharova NV, Tarabukina EB (2017) Synthesis of N-vinylpyrrolidone copolymers with 2-aminoethyl methacrylate as drug carrier. Russ J Gen Chem 87:276–281
Borovikova LN, Titova AV, Kipper AI, Pisarev OA (2017) Complex formation of Daunomycin with poly(vinylpyrrolidone) and poly(ethylene glycol). Russ J Gen Chem 87:1031–1037
Kurmaz SV, Grubenko GA, Knerelman EI, Davydova GI, Torbov VI, Dremova NN (2014) Promising macromolecular nanoobjects for the template synthesis of network copolymers with mesoporous structures. Mendeleev Сommun 24:125–127
Dingcai W, Fei X, Bin S, Ruowen F, He H, Krzysztof M (2011) Design and preparation of porous polymers. Chem Rev 112:3959–4015
Ahmed DS, El-Hiti GA, Yousif E, Ali Ali A, Hameed AS (2018) Design and synthesis of porous polymeric materials and their applications in gas capture and storage: a review. J Polym Res 25:75–95
Nguyen C, Hawker CJ, Miller RD, Huang E, Hendrick JL, Gauderon R, Hilborn JG (2000) Hyperbranched polyesters as nanoporosity templating agents for organosilicates. Macromolecules 33:4281–4284
Hedrick JL, Trollsas M, Hawker CJ, Atthof B, Claesson H, Heise A, Miller RD, Mecerreyes D, Jerome R, Dubois P (1998) Dendrimer−like star block and amphiphilic copolymers by combination of ring opening and atom transfer radical polymerization. Macromolecules 31:8691–8705
Plummer CJG, Garamszegi L, Nguyen TQ, Rodlert M, Manson JAE (2002) Templating porosity in polymethylsilsesquioxane coatings using trimethylsilylated hyperbranched polymers. J Mater Sci 37:4819–4829
Konkolewicz D, Monteiro MJ, Perrier S (2011) Dendritic and hyperbranched polymers from macromolecular units: elegant approaches to the synthesis of functional polymers. Macromolecules 43:5949–5955
Voit BI, Lederer A (2009) Hyperbranched and highly branched polymer architectures – synthetic strategies and major characterization aspects. Chem Rev 109:5924–5973
Tonhauser C, Schüll C, Dingels C, Frey H (2012) Branched acid-degradable, biocompatible polyether copolymers via anionic ring−opening polymerization using an epoxide inimer. ACS Macro Lett 1:1094–1097
Vogt AP, Sumerlin BS (2008) Tuning the temperature response of branched poly(N-isopropylacrylamide) prepared by RAFT polymerization. Macromolecules 41:7368–7373
Wang Z, He J, Tao Y, Yang L, Jiang H, Yang Y (2003) Controlled chain branching by RAFT−based radical polymerization. Macromolecules 36:7446–7452
Malkoch M, Schleicher K, Drockenmuller E, Hawker CJ, Russell TP, Wu P (2005) Structurally diverse dendritic libraries: a highly efficient functionalization approach using click chemistry. Macromolecules 38:3663–3678
Smeets NMB, Freeman MW, McKenna TFL (2011) Polymer architecture control in emulsion polymerization via catalytic chain transfer polymerization. Macromolecules 44:6701–6710
O’Brien N, McKee A, Sherrington DC, Slark AT, Titterton A (2000) Facile, versatile and cost effective route to branched vinyl polymers. Polymer 41:6027–6031
Isaure F, Cormack PAG, Sherrington DC (2004) Facile synthesis of branched poly(methyl methacrylate)s. Macromolecules 37:2096–2105
Slark AT, Sherrington DC, Titterton A, Martin IK (2003) Branched methacrylate copolymers from multifunctional comonomers: the effect of multifunctional monomer functionality on polymer architecture and properties. J Mater Chem 13:2711–2720
Smeets NMB, Moraes RP, Wood J, McKenna TFL (2011) A new method for the preparation of concentrated translucent polymer nanolatexes from emulsion polymerization. Langmuir 27:575–581
Baudry R, Sherrington DC (2006) Facile synthesis of branched poly(vinyl alcohol)s. Macromolecules 39:1455–1460
Chisholm M, Hudson N, Kirtley N, Vilela F, Sherrington DC (2009) Application of the "Strathclyde route" to branched vinyl polymers in suspension polymerization: architectural, thermal, and rheological characterization of the derived branched products. Macromolecules 42:7745–7752
Luzon M, Boyer C, Peinado C, Corrales T, Whittaker M, Tao L, Davis TP (2010) Water−soluble, thermoresponsive, hyperbranched copolymers based on PEG-methacrylates: synthesis, characterization, and LCST behavior. J. Polym. Sci a. Polym Chem 48:2783–2792
Chambon P, Chen L, Furzeland S, Atkins D, Weaver JVM, Adams DJ (2011) Poly(N-isopropylacrylamide) branched polymer nanoparticles. Polym Chem 2:941–949
Besenius P, Slavin S, Vilela F, Sherrington DC (2008) Synthesis and characterization of water-soluble densely branched glycopolymers. React Funct Polym 68:1524–1533
Kurmaz SV, Pyryaev AN (2012) Synthesis and properties of fullerene-containing N-vinylpyrrolidone copolymers. Russ J Gen Chem 82:1705–1714
Kurmaz SV, Pyryaev AN (2010) Synthesis of N-vinyl-2-pyrrolidone-based branched copolymers via crosslinking free-radical copolymerization in the presence of a chain-transfer agent. Polymer Sci Ser B 52:1–8
Gregg SJ, Sing KSW (1982) Adsorption, surface area and porosity. Academic Press, New York
Turner DT, Schartz A (1985) The glass transition temperature of poly(N-vinyl pyrrolidone) by differential scanning calorimetry. Polymer 29:757–762
Deutsche norm Thermische Analyse (2005) DIN 51005. Germany
Zhou Y, Huang W, Liu J, Zhu X, Yan D (2010) Self-assembly of hyperbranched polymers and its biomedical applications. Adv Mater 22:4567–4590
Kurmaz SV, Obraztsova NA, Perepelitsina EO, Shilov GV, Anokhin DV, Pechnikova EV (2015) New hybrid macromolecular structures of C60 fullerene–amphiphilic copolymers of N-vinylpyrrolidone and triethylene glycol dimethacrylate. Mater Today Commun 4:130–140
Saba SA, Mousavi MPS, Buhlmann P, Hillmyer MA (2015) Hierarchically porous polymer monoliths by combining controlled macro- and microphase separation. J Am Chem Soc 137:8896–8899
Acknowledgments
The study was performed in accordance with the government assignment (registry № 01201055328) using the equipment of the Center for Shared Use “Novel Petrochemical Processes, Polymer Composites and Adhesives” (№ 77601).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 3959 kb)
Rights and permissions
About this article

Cite this article
Kurmaz, S.V., Fadeeva, N.V., Knerelman, E.I. et al. Nanoporous polymer networks of N − vinylpyrrolidone with dimethacrylates of various polarity. Synthesis, structure, and properties. J Polym Res 26, 153 (2019). https://doi.org/10.1007/s10965-019-1817-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-019-1817-2