
Abstract
This study synthesized a novel fluoro chain extender, namely 2,2,3,3,4,4,4-heptafluoro-butynic acid 2,2-bis-hydroxymethyl-butyl ester (HFBA), by using 2,2,3,3,4,4,4-heptafluorobutanoyl chloride and trimethylolpropane. Novel polyurethanes (PUs), namely HFBA/PUs, were subsequently synthesized by adopting a hard segment (4,4′-diphenylmethane diisocyanate, MDI), a soft segment (polycaprolactone diol, PCL), and the synthesized chain extender (HFBA). The results of proton nuclear magnetic resonance spectroscopy, fluorine-19 nuclear magnetic resonance, and Fourier transform infrared spectroscopy (FT-IR) demonstrated the successful synthesis of the HFBA chain extender. Gel permeation chromatography revealed that the molecular weight of the HFBA/PUs increased with the HFBA content. Through FR-IR and X-ray photoelectron spectroscopy, we observed a strong hydrogen bond interaction between the NH groups and CF2 or CF3 groups in the HFBA/PUs. This interaction increased with the HFBA content. Additionally, increasing the HFBA content increased the initial decomposition temperature, glass transition temperature, dynamic Tg (Tgd), tensile strength, and Young’s modulus of the HFBA/PUs. These results were because HFBA was a hard segment, which stimulated a stronger interaction between the NH groups and CF2 or CF3 groups in the PUs. By contrast, the HFBA/PUs had low elongation-at-break values. Atomic force microscopy revealed a higher number of bump-like protrusions and higher surface roughness levels among HFBA/PUs with higher HFBA content ratios. Finally, we coated the HFBA/PUs onto polyethylene terephthalate fabrics and discovered that the coated fabrics demonstrated high waterproofing and water vapor permeability levels.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Fluoropolymers are macromolecules with unique characteristics including high oxygen permeability, hydrolytic stability, thermal stability, and chemical resistance, as well as low permittivity and surface free energy. In addition, they are ideal materials for waterproof devices and oil repellents [1, 2]. Polyurethanes (PUs) are widely used for producing distinct materials such as elastomers, fibers, coatings, and adhesives [3,4,5]. The hardness and flexibility of PUs can be controlled by changing the structure of hard and soft segments [6, 7]. Nevertheless, PUs have disadvantages such as low water resistance, low heat resistance, and poor outdoor durability; these disadvantages limit its applications [8].
Fluoride substances can be added to PUs through functionalization. Therefore, the properties of functionalized PUs are similar to those of fluoropolymers [9, 10], such as favorable hydrolytic, thermal, and oxidation stability. Fluorinated PUs (FPUs) also exhibit improved oxygen permeability [11, 12]. Additionally, the surface adhesion FPUs decreases when this material is in direct contact with foreign substances [13]. To add fluorine to functionalized PUs, researchers have mainly adopted three methods, namely soft segment fluorination [14, 15], hard segment fluorination, and chain extender fluorination [16,17,18,19]. Moreover, researchers have synthesized PUs with unique properties by adding fluoride to the polymer [20, 21]. For example, Tao et al. [14] synthesized a series of fluorinated thermoplastic PU elastomers (FTPUs) based on a fluorinated polyether diol. To improve the mechanical properties and molecular weight of the FTPUs, they explored the effectiveness of FTPU compounded with different percentages of polybutylene adipate (PBA). The results confirmed that PBA helped enhance the molecular weight and tensile strength of FTPU; furthermore, FTPUs with higher fluorine content exhibited stronger thermal resistance when compared with other samples. Tonelli [15] synthesized an FPU by using perfluoropolyether as the soft segment; the synthesized polymer exhibited an extremely low friction coefficient, high thermal stability, and excellent chemical resistance. In addition, Su et al. [18] synthesized 2,2,3,3,4,4,5,5-octafluoro-N-(1,3-dihydroxy-2-methylpropan-2-yl) pentanamide (ODMP)—a chain extender—and incorporated it into PU sidechains. Their findings revealed that the fluorinated chain extender increased the thermal properties and tensile strength of the PU; moreover, erythrocyte test results showed that the chain extender reduced the average number of red blood cells that adhered to the polymer surface. Wu et al. [19] synthesized FPUs and PUs by using the chain extenders 2,2,3,3,4,4,5,5,-octafluoro-1,6-hexanediol (OFHD) and 1,6-hexanediol (HD), respectively; they reported that the FPUs were superior to the PUs in terms of thermal and mechanical properties. Su et al. [16] synthesized an FPU by adopting 4-(1,1,1,3,3,3-hexafluoro-2-(4-hydroxyphenyl)propan-2-yl)phenol (HFP); they subsequently conducted an in vitro erythrocyte adhesion test. Their results indicated that increasing the fluorine content (namely HFP content) enhanced HFP/PU resistance to thrombosis. They thus considered the polymer to be feasible for biomedical applications. Massa [20] prepared an FPU to improve the fundamental ecological stability and blood compatibility of PUs.
Waterproof and breathable textiles (WBTs) have been commonly employed in hazmat suit and sportswear applications. However, the waterproofing level of such textiles is inversely proportional to the levels of vapor permeability and breathability. Specifically, loosely woven textile fabrics exhibit high air permeability and low water resistance, and the reverse is the case for tightly woven textile fabrics [22]. The comfort level of WBTs, which are characterized by water resistance and vapor permeability, is also highly essential to users. WBTs can be produced using three types of materials: waterproof fabrics that prevent liquid, vapor, or gas from penetration; waterproof coatings that resist liquid splash or gas/vapor penetration; and waterproof and breathable fabrics that prevent liquid phase or gas/vapor penetration [23]. Waterproof fabrics with low vapor permeability are likely to engender sweating under high-humidity conditions, leading to high body temperatures [24]. Therefore, developing WBTs that can resist water with adequate air permeability levels is necessary. Yun [25] prepared a coating material by blending waterborne PU–urea (WBPU) and sodium alginate (SA); the study results demonstrated that the waterproofing level decreased as the SA content increased. Kwak et al. [26] selected polyols poly(tetramethylene oxide) glycol (PTMG, polypropylene glycol (PPG), polycaprolactone (PCL), and poly(tetramethylene adipate glycol) PTAd), polyol blends (PCL/PPG, PCL/PTMG, PTAd/PPG, and PTAd/PTMG), and triblock diol [(CL)4.5-PTMG-(CL)4.5 (TBG) and PCL–PEG–PCL) to study the water vapor permeability (WVP) of coatings. Moreover, the waterproofing level of WBPU can be adjusted by changing soft segment types and molecular chains. Meng et al. [29] fabricated WBPU by using ethylene oxide–propylene oxide (EO–PO) copolymers and polytetramethylene ether glycol (PTMEG) mixture. They proved that PUs prepared using PPG2050/PTMEG were superior to those prepared using PEG/PPG/PTMEG in terms of water resistance.
In this study, we synthesized a new fluorinated chain extender, namely 2,2,3,3,4,4,4-heptafluoro-butynic acid 2,2-bis-hydroxymethyl-butyl ester (HFBA). This extender was synthesized through the esterification of 2,2,3,3,4,4,4-heptafluorobutanoyl chloride (HFC) and trimethylolpropane (TMP). Next, a prepolymer was obtained by reacting 4,4′-diphenylmethane diisocyanates (MDI) and PCL diol. The HFBA extender was then added to the prepolymer, thus yielding HFBA/PU polymers. Finally, we examined the effects of adjusting the content of PCL and the HFBA chain extender on the structure, molecular weight, thermal properties, mechanical properties, and surface properties of the HFBA/PUs. Furthermore, we applied the PUs on polyethylene terephthalate (PET) fabrics and conducted WVP and waterproofing tests.
Experiment
Materials
2,2,3,3,4,4,4-heptafluorobutanoyl chloride (HFC), trimethylolpropane (TMP), triethylamine (TEA), 4,4′-diphenylmethane diisocyanate (MDI), polycaprolactone diol (PCL) (Mw = 530), and dibutyltin dilaurate (DBTDL) were purchased from Aldrich. Tetrahydrofuran (THF) and N,N-dimethylacetamide (DMAc) were obtained from Mallinckrodt Chemicals.
Synthesis of HFBA
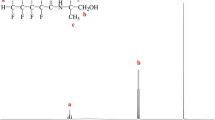
First, HFC and an adequate amount of dry THF were loaded into a 500 mL flat-bottomed flask and then mixed thoroughly at room temperature and under nitrogen flux. TEA (mole ratio: 1.5) was then added to the mixture. Subsequently, dry THF was dissolved using TMP and then injected into the mixture through a syringe. The HFC-to-TMP mole ratio was 1:1. A rotary evaporator was then used to remove the solvent from the mixture, which was washed with ether and deionized water and dried in a vacuum oven at 70 °C for 12 h to obtain the HFBA product. Next, column chromatography was performed to purify HFBA. Finally, vacuum drying and subsequent drying were conducted to dry the purified HFBA in an oven at 50 °C for 1 day. The reaction formula for HFBA is presented in Scheme 1. 1H NMR (DMSO-d6, Fig. 1) δ ppm a: 0.76(m, 3H, Ha), b: 1.20 (m, 2H, Hb), c: 3.26 (d, 4H, Hc), d: 4.20 (m, 2H, Hd). 19F NMR (DMSO-d6, Fig. 2) δ ppm −127.24, −116.84 and − 81.11.

Synthesis formula for the HFBA

1H NMR spectra of the HFBA

19F NMR spectra of the HFBA
Synthesis of HFBA/PUs
MDI, PCL, and DMAc were first placed in a 500 mL three-neck reaction flask and heated to 80 °C. Two to three drops of DBTDL catalyst were subsequently added to the mixture and stirred using a stirrer at 200 rpm. After 2 h of reaction, a PU prepolymer was formed. The HFBA chain extender was dissolved in DMAc and reacted with the prepolymer for 2 h (Scheme 2). The resulting HFBA/PUs were poured into a serum bottle and stored in a refrigerator for 1 day. Finally, the HFBA/PUs were poured over a Teflon plate and dried in temperature-programmable circulating oven for 8 h. Table 1 presents the formula, symbols, hard segment content (Eq. (1)), and soft segment content (Eq. (2)) for the HFBA/PU films.

Synthesis formula for the HFBA/PUs
- W MDI :
-
weight of MDI
- W PCL :
-
weight of PCL
- W HFBA :
-
weight of HFBA
Gel permeation chromatography
Gel permeation chromatography (GPC) was conducted using the Analytical Scientific Instrument Model 500 system. A refractive index detector (Schambeck RI2000), two mixed-bed Jordi gel DVB columns, and a 10,000 Å bed at 30 °C were used to measure the molecular weight distribution relative to polystyrene standards. Eight analytical standards were applied to calibrate the molecular weight from 3420 to 2.57 × 106. The solvent was tetrahydrofuran applied at a flow rate of 1 mL/min.
1H NMR spectrometry
The 1H NMR (in DMSO-d6) spectra of the specimens were measured using a Bruker Avance 300 spectrometer (300 MHz).
19F NMR spectrometry
The 19F NMR spectra of the polymers were recorded using a Bruker Avance IIIHD 400 Hz spectrometer. DMSO-d6 served as the solvent and tetramethylsilane served as the internal standard.
Fourier transform infrared spectroscopy
Fourier transform infrared spectroscopy (FT-IR) was conducted using a PerkinElmer spectrometer (model Spectrum One). FT-IR measurements were conducted at bands ranging from 4000 to 650 cm−1, and scanning was conducted 16 times on average at a resolution of 2 cm−1.
X-ray photoelectron spectroscopy
X-ray photoelectron spectroscopy (XPS) was conducted using a Thermo Fisher Scientific(VGS)spectrometer. An Al Kα anode was adopted as the X-ray source (1486.6 eV). Binding energies ranging from 0 to 1400 eV were selected for analysis. The binding energies were calibrated to the C1s internal standard with a peak at 284.8 eV. High-resolution C1s spectra were decomposed by fitting a Gaussian function to an experimental curve by using a nonlinear regression approach.
Thermogravimetric analysis
Thermogravimetric analysis (TGA) was conducted using a PerkinElmer TGA instrument (model Pyris 1). At room temperature, specimens weighing 5–8 mg were heated to 700 °C under nitrogen flux at a rate of 10 °C/min.
Differential scanning calorimetry
Differential scanning calorimetry (DSC) was conducted using a PerkinElmer DSC instrument (model Jade). Specimens were sealed in aluminum pans and heated from −50 to 50 °C under nitrogen flux at a rate of 10 °C/min. The glass transition temperature (Tg) was determined at the midpoint of the DSC curve. All specimens weighed 5–8 mg.
Dynamic mechanical analysis
Dynamic mechanical analysis was conducted using a SEIKO DMS instrument (model SII Muse, DMS6100). The measurement was conducted in tension mode using the following parameters: frequency, 1 Hz; amplitude, 5 μm; and temperature range, −50 to 50 °C. Moreover, the heating rate was 3 °C/min. The specimens measured 20 (L) × 5 (W) × 0.2 (H) mm3. Tg was considered as the peak temperature in the glass transition region in the tanδ curve.
Stress–strain testing
A universal testing machine (MTS QTEST5, model QC505B1) was used to measure the specimens’ tensile strength and elongation at break. Testing was conducted according to ASTM D638. The coating specimens measured 45 × 8 × 0.2 mm3.
Contact angle analysis
A face instrument (model CA-VP150) was used to measure the contact angles of deionized water droplets on specimen surfaces at room temperature. Droplets were deposited from a syringe pump onto the specimen surface. The corresponding contact angles were then recorded. Each reported contact angle is the mean of contact angles measured for three to four droplets.
Surface roughness analysis
An atomic force microscope (model CSPM5500, manufactured by Being Nano-Instruments) was used to scan the surface roughness of the specimens. In generally, tapping and contact modes are used for analysis. The present study adopted the tapping mode, with the probe indirectly contacting the specimen surface using a cantilever. For the sine-wave phases of the driving cantilever, the tip oscillation phases were determined to be sensitive to the surface characteristics of the specimens. Therefore, except for topography, the tip could capture phase images of the specimen surfaces. The testing specimens were HFBA/PU films with distinct HFBA content levels.
Stain resistance
PUs with distinct HFBA ratios were applied to PET fibers through an automatic applicator. Water, black tea, and blue ink were dropped from pipettes onto the PET fibers with HFBA/PU coatings to determine their stain resistance. Next, the droplet conditions were photographed and observed. PET without HFBA/PU was also included in the experiment for comparison.
Fabricating the HFBA/PU films and fabric coating
PUs with distinct HFBA ratios were padded on PET fabrics by using a padding mangle; this process was executed using a “two-dips, two-nips” method under a pressure of 1 Kg/m2. Subsequently, the fabrics were dried at 60 °C for 10 min and then at 90 °C for 20 min. The WVP of the fabrics was then measured. WVP (g/m2/24 h), determined at a specific temperature and moisture level, is a measure of the time required for water vapor to pass through a material’s surface. In general, PU-coated fabrics must be integrated with microporous films or nonporous hydrophilic films to ensure that they become permeable to water vapor while remaining waterproof. When a microporous film is applied, the fabric becomes impermeable to water but permeable to water vapor. When a nonporous hydrophilic film is applied to the fabric, vapor can be absorbed through the hydrophilic region of the fabric coating, proliferation, and desorption. Waterproofing tests were conducted according to the JIS L1099 standard for high-water-pressure tests. Regarding spraying tests, ratings are categorized into 69 ISO levels: level 0 (0 point), indicating complete wetting of the surface and back; level 1 (50 points), indicating complete wetting of the surface; level 2 (70 points), indicating partial wetting; level 3 (80 points), indicating wetting of the surface at spray points; level 4 (90 points), indicating a dry surface with spray points; and level 5 (100 points), indicating a dry surface without spray points. The coated fabrics were tested after they were washed in water (40 °C) five times.
Results and discussion
GPC analysis
Figure 3 illustrates the GPC curves of HFBA/PUs with distinct HFBA ratios, indicating unimodal molecular weight distributions of the synthesized PUs with no redundant raw materials. Table 2 presents the weight-average molecular weight (Mw) and number-average molecular weight (Mn) of the specimens. According to the experimental data, the molecular weights of HFBA/PUs increased with the HFBA content. There is a certain relationship between molecular weight and mechanical properties. The PU which had larger molecular weight will generally show better mechanical properties as will be shown in the following related sections. The molecular weight distributions or polydispersities (PDIs) were determined as the Mw/Mn ratio, which ranged from 2.1 to 1.4, signifying that as the HFBA content was increased, the molecular weight distributions became narrower. The variation trend of PDI with chain extender content is similar to the result reported by Zhu et al. [27]. The reason for this behavior is not easy to be identified. One possibility is that the higher degree of microphase separation causes the narrower distribution of molecular weight of PUs [27]. On the other hand, it is well known that the degree of microphase separation increases with the increasing of hard segment content, i.e. HFBA content. So the molecular weight distribution became narrower as the HFBA content was increased.

GPC curves of the HFBA/PUs
FT-IR spectroscopy
The FT-IR spectrum of the HFBA chain extender is presented in Fig. 4a. As indicated in the figure, an absorption peak at 3323 cm−1 was observed and was assigned to the hydroxyl group (-OH). Symmetric and asymmetric CH2 stretching vibration peaks appeared at 2928 and 2849 cm−1, respectively, and were attributed to the original trimethylolpropane molecule. Moreover, strong absorption peaks were noted at 881 and 1222 cm−1 and were attributed to the stretching vibration of CF3 and CF2, respectively. The peak at 1623 cm−1 was ascribed to the carbonyl group (C=O). Both Figs. 1 and 4a verify the successful synthesis of HFBA in this study.

FT-IR spectra of the HFBA and HFBA/PUs at the wavenumber range of (a), b 4000-650 cm−1; c 1900-1200 cm−1
Figure 4b illustrates the FT-IR spectra of the HFBA/PUs at wavenumbers ranging from 4000 to 650 cm−1. As indicated in the spectra, five common peaks attributed to macromolecules were observed. A peak was noted at 3310 cm−1 and was ascribed to the stretching vibration of urethane NH group. Furthermore, peaks appeared at 2944 and 2868 cm−1 and were assigned to CH2 stretching. And peaks at 1727 and 1712 cm−1 were attributed to the urethane C=O group. Additionally, a peak was determined at 1532 cm−1 and was attributed to the NH vibration of associated secondary urethane groups. Moreover, stretching vibration peaks appearing between 1641 and 1592 cm−1 were ascribed to the C=O group connected with C-F group. The bands at between 1641 and 1592 cm−1 and at between 1727 and 1712 cm−1 show two types of different C=O groups, namely, the C=O connected with C-F and the C=O in usual urethane. A stretching vibration peak was observed at 1270 cm−1 and was assigned to the C–F group, and another at approximately 1219 cm−1 was attributed to the C–O group. In addition, no free NCO group was detected between 2240 and 2275 cm−1. Consequently, the spectra indicated that MDI was completely reacted with PCL and HFBA during the synthesis processes.
Figure 4c shows the absorption peaks at wavenumbers ranging between 1900 and 1200 cm−1. The urethane NH deformation vibration (amide II band) peaks of HFBA/PU-01, HFBA/PU-02 and HFBA/PU-03 were located at 1511 cm−1, 1520 and 1532 cm−1, respectively. So the urethane NH vibration peak of a HFBA/PU with higher HFBA content shifted to higher wavenumber. This is due to the interaction between NH and CF2 (or CF3) groups. The interaction is more profound when more CF2 (or CF3) group or higher HFBA content is present as the H-bonding interactions behavior reported in literature [28]. The urethane C=O stretching of the HFBA/PUs were noted at 1727 and 1712 cm−1 for the free and interacted (or hydrogen-bonded) C=O stretchings, respectively. For the HFBA/PU-01, the peak at 1712 cm−1 was smaller than that at 1727 cm−1. But the relative magnitude of the peak at 1712 cm−1 to that at 1727 cm−1 increased with increasing HFBA content, and the peak at 1712 cm−1 was larger than that at 1727 cm−1 for HFBA/PU-03. This is because the C=O group is more interacted by more CF2 (or CF3) groups in HFBA/PU-03, similar to the behavior reported by Wu et al. [19]. The influence of C=O stretching by the interaction between C=O group and CF2 (or CF3) group is similar to the H-bonding interaction between C=O and NH groups reported in literature [30, 31].
From the previous discussion, HFBA and PCL had reacted with MDI to form HFBA/PUs because of the following three deductions. First, the C=O stretching at 1623 cm−1 in HFBA was shifted to 1641 cm−1 in HFBA/PUs resulting from hydrogen bonding interaction in the polymeric system. Second, no free NCO group was detected in HFBA/PUs, indicating that MDI was completely reacted with HFBA and PCL according to the formulas in Table 1 during the synthesis processes. Third, the characteristic peaks of polyurethanes were observed, i.e. urethane –NH stretching at 3310 cm−1, urethane C=O stretching at 1727 and 1712 cm−1, and urethane -NH deformation from 1532 to 1511 cm−1 (amide II band).
19F NMR
Figure 5 is the 19F NMR spectrum of HFBA/PU-03. Three absorption peaks are observed at −127.12, −116.78 and − 81.07 ppm. The three F valences were correspondingly assigned as 1–3. Located between −110 and − 140 ppm, two peaks are consistent with the typical range of CF2 signals [32,33,34]. One peak are consistent with the typical range of CF3 signals [35].

19F NMR spectrum of the HFBA/PU-03
To accurately assign absorption peaks 1–3 to the F valences of the molecular formula of HFBA/PU as F1, F2 and F3, the 19F–19F correlation spectrum of HFBA/PU-03 was examined (Fig. 6). Three instances of strong couplings between next-nearest neighbors (i.e., F2–F3 [A], and F1–F2 [B]) were observed. Thus, the peaks located at −127.12, −116.78, and − 81.07 ppm corresponded to F1, F2, and F3, respectively. Hence, the HFBA extenders successfully formed urethane groups with the MDI monomers.

19F–19F COSY NMR spectrum of the HFBA/PU-03
XPS analysis
XPS was conducted to determine the elemental compositions and chemical state of the synthesized HFBA/PUs. Figure 7 illustrates the spectra of these macromolecules. As indicated in this figure, four peaks were observed for each HFBA/PU specimen, corresponding to C1s, O1s, F1 s and N1 s orbits, respectively. Moreover, another peak corresponding to the Sn3d orbit was attributed to the presence of dibutyltin dilaurate used as a catalyst in the synthesis processes of HFBA/PUs.

XPS survey spectra of the HFBA/PUs
Table 3 presents the elemental compositions and associated peak characteristics. As shown in this table, the fluorine content in the HFBA/PUs increased with the HFBA content. The measured C1s binding energies of HFBA/PU-01, HFBA/PU-02, and HFBA/PU-03 were 283.85, 284.20, and 284.77 eV, respectively. Moreover, the measured F1 s binding energies in the C-F bonds of HFBA/PU-01, HFBA/PU-02, and HFBA/PU-03 were 687.55, 687.28, and 687.36 eV, respectively. Previous studies [36, 37] have indicated that the F1 s peak of fluorinated copolymers containing distinct neighborhoods could be presented as a single fitable component; by contrast, the C1s peak required the same number of components as the number of neighborhoods. However, in the present study, fluorine demonstrated different binding energies in distinct polymers. The C1s binding energy of the CH2 group and F1 s binding energy of the CF group in the polymer (–CFCF3CF2 –)n(–CF2CH2 –)n were 286.77 and 688.8 eV, respectively. The C1s binding energy of the CH2 group and F1 s binding energy of the CF group in the (–CHFCH2–)n were 285.74 and 686.94 eV, respectively. Furthermore, the C1s binding energy of the CH2 group and F1 s binding energy of the CF group in the polymer (–CHFCH2–)n were 292.48 and 689.67 eV, respectively. The C1s binding energy of the CH2 group and F1 s binding energy of the CF group in the polymer (–CF2CF2–)n were 292.48 and 689.67 eV, respectively. The results thus confirmed that the lower C1s and F1 s binding energies were engendered by the interaction between F and CH. Similarly, the results of this study demonstrated that for (–CF2CF2–)n, the C1s binding energy of the CH2 group (292.48 eV) were lower than the F1 s binding energies (689.67 eV) of the C–F bonds [36]. When C1s and F1 s binding energies declined, the mentioned effects proved the interaction between the NH and CF2 or CF3 groups of the HFBA/PUs. Furthermore, the surface nitrogen content increased with the HFBA content possibly because the hydrogen bond interaction occurred among the protons in the C–F groups of HFBA and those in the N–H groups of urethane [38]. Therefore, the present study determined that portions of hard segments can exposed on the surface by using fluorinated sidechains [39]. The presented FT-IR results provide relevant evidence.
Thermal properties
Figure 8 presents the TGA and DTG curves of the HFBA/PUs with distinct ratios of the HFBA chain extender. Only one decomposition temperature was identified from the curves, which was attributed to the decomposition of the urethane group and was thus named Tonset. The onset temperatures were the initial decomposition temperatures of these polymers. The initial decomposition temperatures of HFBA/PU-01, HFBA/PU-02, and HFBA/PU-03 were 326.2, 338.4, and 349.3 °C, respectively (Table 4). Therefore, the initial decomposition temperatures increased with the content of HFBA and hard segments. In other words, elevating the content of HFBA and hard segments improved the thermal resistance of the polymers [40]. Su et al. [18] synthesized fluorinated ODMP/PUs with the same fluorinated chain extender, but the initial decomposition temperature is lower than that in the present study, probably because the bonding energy of the C–O group in HFBA (358 kJ/mol) is stronger than that of the C–N group in ODMP (284 kJ/mol). Moreover, the sidechains of HFBA were shorter than those of ODMP. Increasing the HFBA content boosted the initial decomposition temperatures of the HFBA/PUs, which was due to the strong interactions between the –NH and CF2 or CF3 groups in the HFBA/PUs, as observed in the FT-IR and XPS spectra.

TGA and DTG curves of the HFBA/PUs
Figure 9 presents DSC thermograms of HFBA/PUs with different HFBA content levels, and the DSC results are summarized in Table 4. The Tg values of HFBA/PU-01, HFBA/PU-02, and HFBA/PU-03 were − 3.8, 2.9, and 5.7 °C, respectively. The results indicate that increasing the HFBA content reduced the PCL content, thus increasing the Tg values of the PUs. Similarly, increasing the content of hard segments or HFBA increased the Tg values of the PUs. By contrast, increasing the soft segment content reduced the Tg values of the PUs. Moreover, large sidechains were key factors influencing the movement of molecular chains. Compared with the results reported by Su et al. [18], the present study identified that the Tg value of the ODMP/PU was lower than that of the HFBA/PU, although they had the same amount of chain extenders. Consequently, for molecules with longer sidechains, the free volume of molecular chains was expanded, further reducing Tg. Furthermore, the FT-IR and XPS spectra confirmed that a strong interaction existed between the –NH and CF2 or CF3 groups. The interaction hindered segmental motion of the HFBA/PUs, increasing the Tg values of the HFBA/PUs. These results were determined to be similar to the TGA results: increasing the HFBA content increased the initial decomposition temperature of the PUs.

DSC thermograms of the HFBA/PUs
Dynamic mechanical analysis
Figure 10 shows the tanδ and loss modulus (E”) values of the HFBA/PUs with different HFBA ratios. Tgd is typically determined as the peak temperature of tanδ or E “curves. According to the tanδ curves, the Tgd values of HFBA/PU-01, HFBA/PU-02, and HFBA/PU-03 were 7.0, 15.1, and 20.4 °C, respectively. Whereas the Tgd values of HFBA/PU-01, HFBA/PU-02, and HFBA/PU-03 were 2.5, 7.4, and 15.6 °C respectively, according to the E” curves. Different measurements gave different Tgd values. The whole results are presented in Table 5. The hard segments are more rigid and less easy to undergo segmental motion than the soft segments, so the Tgd values of the HFBA/PUs increased with the HFBA content or the hard segment content. Furthermore, the CF2 or CF3 groups in the HFBA will interact with the -NH groups in PUs to hinder the segmental motion in the HFBA/PUs and raise the Tgd. This behavior of the Tgd values of HFBA/PUs increasing with the HFBA content is similar to the DSC results.

Tan δ and loss modulus (E”) curves of the HFBA/PUs
As indicated in the tanδ curves, the Tgd peak value of a HFBA/PU increases with lower HFBA or hard segment content. The tanδ is associated with the viscoelastic property of the polymer and is equals to E“/E’. The loss modulus E” is a damping or energy dissipation term and the storage modulus E’ is associated with the elastic behavior. The HFBA/PU with higher soft segment has higher damping behavior and lower elastic chateristics, so the HFBA/PU with higher soft segment or lower HFBA content has a higher Tgd peak value.
Tensile properties
Figure 11 presents stress–strain curves of the HFBA/PUs with distinct HFBA ratios. These curves correspond to the mechanical property data listed in Table 6. The measured tensile strength levels of HFBA/PU-01, HFBA/PU-02, and HFBA/PU-03 were 10.2, 16.5, 20.9 MPa, respectively, and their Young’s moduli were 0.8, 2.1, and 4.5 MPa, respectively. The results indicated that when the HFBA (hard segment) content increased in the PUs, the polymer stiffness increased, thus increasing the tensile strength and Young’s modulus. Additionally, the strong interaction between the NH and CF2 or CF3 groups in the HFBA/PUs hindered the segmental motion of the HFBA/PUs, consequently increasing the tensile strength and Young’s modulus of the PUs. The elongation at break of HFBA/PU-01, HFBA/PU-02, and HFBA/PU-03 were 861%, 574%, and 391% respectively. We thus inferred that increasing the HFBA or hard segment content resulted in unfavorable sliding motions among polymer chains. Therefore, HFBA/PUs with more HFBA demonstrated lower elongation-at-break values. Wu et al. [19] also synthesized PUs with fluorine as the main chain; they reported that the tensile strength and elongation at break of the synthesized PUs were higher than those of HFBA/PUs, possibly because the sidechain groups in macromolecules tend to disrupt the molecular distributions. Therefore, reducing the tensile strength of macromolecules results in unfavorable sliding motions among polymer chains.

Tensile properties of the HFBA/PUs
Contact angle analysis
Figure 12 illustrates the surface activity of the HFBA/PUs determined according to their wetting ability. The contact angles between water molecules and the surfaces of HFBA/PU-01, HFBA/PU-02, and HFBA/PU-03 were 95.4°, 101.0°, and 104.4°, respectively. These results indicate that the contact angles increased with the HFBA content. In principle, the level of polymer surface hydrophilicity or hydrophobicity is largely determined by chemical compositions. When the HFBA content is high, more CF2 or CF3 groups are attached to PUs. The surface tension and hydrophilicity of such groups are lower than those of other functional groups [41, 42]. Therefore, HFBA/PUs with high HFBA content can be considered to be more hydrophobic with greater contact angles.

Surface properties of the HFBA/PUs
Stain resistance
Figure 13 depicts the pictures obtained from measuring the resistance of HFBA/PU-coated PET fabrics against staining by water, black tea, and blue ink. A PET fabric without HFBA/PU coating was not resistant against any of the mentioned liquids. Specifically, the liquids quickly penetrated the fabric. By contrast, when water (transparent) and black tea (yellow) were dripped on the three PET fabrics coated with different HFBA/PU ratios, each droplet assumed a spherical shape. As revealed in Fig. 13c, blue ink droplets penertrated the HFBA/PU surface to a greater extent compared with droplets of the other two liquids. Blue ink is more likely to attach to the HFBA/PU surface probably because it contains organic solvents or auxiliary substances. However, when the HFBA content increased, the sphericity of all droplets further increased. These results were determined to be consistent with those obtained from the contact angle analysis.

Stain resistance of the HFBA/PUs
Surface roughness analysis
Figure 14 presents the topography (left) and phase data (right) of the specimens. According to the topography data, all HFBA/PUs exhibited successive bump-like protrusions. In particular, when the HFBA content was high, the protrusions tended to greater. The average surface roughness levels of HFBA/PU-01, HFBA/PU-02, and HFBA/PU-03 were 4.16, 12.2, and 27.0 nm, respectively. Similarly, increasing the HFBA content increased the surface roughness levels of the PUs. The observed protrusions were attributed to the hard-segment-rich domains of the PUs or to the high HFBA content. The CF2 or CF3 groups were noted to demonstrate a strong interaction with the NH groups in hard-segment-rich domains, consequently leading to surface aggregations or protrusions [18]. Furthermore, the phase data of the fluorinated polymers (HFBA/PUs) indicated a high number of phases involving irregular particles and stripes. Increasing the HFBA content of the HFBA/PUs increased the number of phases involving irregular particles and stripes. This phenomenon indicates that the hard segments in this study were rich of HFBA chain extenders [43, 44].

AFM topographic and phase images of the HFBA/PUs for (a) HFBA/PU-01, (b) HFBA/PU-02, and (c) HFBA/PU-03
Analyses of WVP and waterproofing levels of HFBA/PU-coated fabrics
Table 7 presents the WVP values of PET fabrics coated with different HFBA/PU ratios. The results revealed that the WVP values of coated fabrics were lower than that of the uncoated fabric. The values also decreased as the HFBA content increased. Each specimen passed the waterproofing test, with the corresponding waterproofing levels exceeding 10,000 mmH2O, thus demonstrating the ability of the fabrics to resist rain or rain drops (whose diameter ranges from 100 to 600 μm). Additionally, the HFBA/PU-coated PET fabrics were determined to meet ISO standards, achieving 90 points. According to these results, we can conclude that we successfully developed HFBA/PU coatings exhibiting high waterproofing and WVP levels.
Conclusions
The study was based on the concepts of molecular geometry, organic chemistry, and macromolecular synthesis. We inserted CF2 groups into PUs and prepared novel HFBA/PUs. Analyses of the molecular geometry of HFBA and the HFBA/PUs were conducted through 1H NMR, XPS, and FT-IR spectroscopy. The results reveal a strong interaction between the NH groups and CF2 or CF3 groups. The initial decomposition temperature, Tg, Tgd results, tensile strength, and Young’s modulus all increased with the HFBA content. Moreover, increasing the HFBA content enhanced the hydrophobicity of the HFBA/PUs. HFBA/PUs with more HFBA chain extenders exhibited higher waterproofing levels. All HFBA/PU-coated PET fabrics met IOS level 4 and above.
References
Kawakami H, Mikawa M, Takagi J, Nagaoka S (1996) Gas transfer and blood compatibility of fluorinated polyimide membranes. J Biomater Sci Polym Ed 7:1029–1038
Tonelli C, Trombetta T, Scicchitano M, Castiglioni G (1995) New perfluoropolyether soft segment containing polyurethanes. J Appl Polym Sci 57:1031–1042
Hirose M, Kadowaki F, Zhou JH (1997) The structure and properties of core–shell type acrylic–polyurethane hybrid aqueous emulsions. Prog Org Coat 31:157–169
Tien YI, Wei KH (2001) Hydrogen bonding and mechanical properties in segmented montmorillonite/polyurethane nanocomposites of different hard segment ratios. Polymer 42:3213–3221
Mequanint K, Sanderson R (2003) Synthesis and crosslinking with melamine formaldehyde resin. Orig Polym 44:2631–2639
Kojio K, Nakashima S, Furukawa M (2007) Microphase-separated structure and mechanical properties of norbornane diisocyanate-based polyurethanes. Polymer 48:997–1004
Oprea S (2007) Synthesis and characterization of polyurethane urea acrylates: effects of the hard segments structure. J Appl Polym Sci 105:2509–2515
Luo Q, Shen Y, Li P, Wang C, Zhao Z (2014) Synthesis and characterization of crosslinking waterborne fluorinated polyurethane-acrylate with core-shell structure. J Appl Polym Sci 131:8558–8572
Baradie B, Shoichet MS (2005) Novel fluoro-terpolymers for coatings applications. Macromolecules 38:5560–5568
Scheirs J (1997) Modern fluoropolymers. Wiley, New York
Ho T, Wynne KJ (1992) A new fluorinated polyurethane: polymerization, characterization, and mechanical properties. Macromolecules 25:3521–3527
Xiong SD, Guo XL, Li L, Wu SL, Chu PK, Xu ZS (2010) Preparation and characterization of fluorinated acrylate copolymer latexes by miniemulsion polymerization under microwave irradiation. J Fluor Chem 131:417–425
Jiang M, Zhao XL, Ding XB, Zheng ZH, Peng YX (2005) A novel approach to fluorinated polyurethane by macromonomer copolymerization. Eur Polym J 41:1798–1803
Tao L, Lin Y (2010) Synthesis and properties of fluorinated thermoplastic polyurethane elastomer. J Fluor Chem 131:36–41
Tonelli C, Ajroldi G (2003) New fluoro-modified thermoplastic polyurethanes. J Appl Polym Sci 87:2279–2294
Su SK, Gu JH, Lee HT, Yu SH, Wu CL, Suen MC (2016) Effects of an aromatic Fluoro-diol and Polycaprolactone on the properties of the resultant polyurethanes. Adv Polym Technol 37:1142–1152. https://doi.org/10.1002/adv.21773
Chen KY, Kuo JF (2000) Synthesis and properties of novel fluorinated aliphatic polyurethanes with fluoro chain extenders. Macromol Chem Phys 201:2676–2686
Su SK, Gu JH, Lee HT, Wu CL, Hwang JJ, Suen MC (2017) Synthesis and properties of novel biodegradable polyurethanes containing fluorinated aliphatic side chains. J Polym Res 24:142–160
Wu CL, Chiu SH, Lee HT, Suen MC (2016) Synthesis and properties of biodegradable polycaprolactone/polyurethanes using fluoro chain extenders. Polym Adv Technol 27:665–676
Massa TM, McClung WG, Yang ML, Ho JYC, Brash JL, Santerre JP (2007) Fibrinogen adsorption and platelet lysis characterization of fluorinated surface modified polyetherurethanes. J Biomed Mater Res 81A:178–185
Jahangir AR, McClung WG, Cornelius RM, McCloskey CB, Brash JL, Santerre JP (2002) Fluorinated surface-modifying macromolecules: modulating adhesive protein and platelet interactions on a polyether-urethane. J Biomed Mater Res 60:135–147
McCord MG (2000) (2000 ) in performance of protective clothing: issues and priorities for the 21st century. In: Nelson CN, Henry NW (eds). American Society for Testing and Materials, PA, p 87
Hao X, Zhang J, Guo Y (2004) Study of new protective clothing against SARS using semi-permeable PTFE/PU membrane. Eur Polym J 40:673–678
Lee S, Obendorf SK (2006) Developing protective textile materials as barriers to liquid penetration using melt-electrospinning. J Appl Polym Sci 102:3430–3437
Yun JK, Yoo HJ, Kim HD (2007) Preparation and properties of waterborne polyurethane-urea/sodium alginate blends for high water vapor permeable coating materials. J Appl Polym Sci 105:1168–1176
Kwak YS, Park SW, Lee YH, Kim HD (2003) Preparation and properties of waterborne polyurethanes for water-vapor-permeable coating materials. J Appl Polym Sci 89:123–129
Zhu R, Wang X, Yang J, Wang Y, Zhang Z, Hou Y, Lin F, Li Y (2017) Influence of hard segments on the thermal, phase-separated morphology, mechanical, and biological properties of polycarbonate urethanes. Appl Sci 2017(7):306. https://doi.org/10.3390/app7030306
Zhang C, Ren Z, Yin Z, Qian H, Ma D (2008) Amide ii and amide iii bands in polyurethane model soft and hard segments. Polym Bull 60:97–101
Meng QB, Lee SI, Nah C, Lee YS (2009) Preparation of waterborne polyurethanes using an amphiphilic diol for breathable waterproof textile coatings. Prog Org Coat 66:382–386
Brunette CM, Hsu SL, Macknight WJ (1982) Hydrogen-bonding properties of hard-segment model compounds in polyurethane block copolymers. Macromolecules 15:71–77
Chen SJ, J l H, Zhuo HT, Yuen CW, Chan LK (2010) Study on the thermal-induced shape memory effect of pyridine containing supramolecular polyurethane. Polymer 51:240–248
Mohanty AK, Misra M, Drzal LT (2002) Sustainable biocomposites from renewable resources: opportunities and challenges in the green materials world. J Polym Environ 10:19–26
Gunatillake P, Mayadunne R, Adhikari R, El-Gewely MR (2006) Recent developments in biodegradable synthetic polymers. Biotechnol Annu Rev 12:301–347
Pena J, Corrales T, Izquierdo-Barba I, Doadrio AL, Vallet-Regi M (2006) Long term degradation of poly(ε-caprolactone) films in biologically related fluids. Polym Degrad Stab 91:1424–1432
Scott Prosser R, Luchette PA, Westerman PW (2000) Using O2 to probe membrane immersion depth by 19F NMR. Proc Natl Acad Sci 97:9967–9971
Beamson G, Briggs D (1992) High resolution XPS of organic polymers:the scienta ESCA300 database. Wiley, New York
Ferraria AM, Silva JDL, Rego AMB (2003) XPS studies of directly fluorinated HDPE: problems and solutions. Polymer 44:7241–7249
Wang LF (2007) Experimental and theoretical characterization of the morphologies in fluorinated polyurethanes. Polymer 48:894–900
Ge Z, Zhang X, Dai J, Li W, Luo Y (2009) Synthesis, characterization and properties of a novel fluorinated polyurethane. Eur Polym J 45:530–536
Tsou CH, Lee HT, Tsai HA, Cheng HJ, Suen MC (2013) Synthesis and properties of biodegradable polycaprolactone/polyurethanes by using 2,6-pyridinedimethanol as a chain extender. Polym Degrad Stab 98:643–650
Li H, Zhang ZB, Hu CP, Wu SS, Ying SK (2004) Surface composition and property of film prepared with aqueous dispersion of polyurethaneurea-acrylate including fluorinated block copolymer. Eur Polym J 40:2195–2201
Kim YS, Lee JS, Ji Q, McGrath JE (2002) Surface properties of fluorinated oxetane polyol modified polyurethane block copolymers. Polymer 43:7161–7170
Sauer BB, Mclean RS, Thomas RR (1998) Tapping mode AFM studies of Nano-phases on fluorine-containing polyester coatings and Octadecyltrichlorosilane monolayers. Langmuir 14:3045–3051
Brandsch R, Bar G, Whangbo MH (1997) On the factors affecting the contrast of height and phase images in tapping mode atomic force microscopy. Langmuir 13:6349–6353
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Su, SK., Gu, JH., Lee, HT. et al. Biodegradable polyurethanes: novel effects of the fluorine-containing chain extender on the thermal, physical and water vapor permeation properties. J Polym Res 25, 227 (2018). https://doi.org/10.1007/s10965-018-1607-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-018-1607-2