
Abstract
In this study polypropylene/ethylene-propylene rubber (PP/EPR) and different amounts of partially reduced graphene oxide (prGO) and its derivatives were investigated. The nanocomposites were also compatibilized with EPR containing diethyl maleate (DEM), glycidyl methacrylate (GMA) and maleic anhydride (MAH) functionality as a means of controlling their ultimate mechanical behaviour and their structural morphology. Mechanical tests show that the addition of pristine prGO to PP/EPR blend promotes tensile strength and Young’s modulus, while reducing elongation at break and impact strength. A significant improvement of these properties was achieved by the presence of functionalized prGO and EPR-g-DEM. TGA analysis showed that the nanocomposites exhibit a higher thermal stability than that of the matrix alone. XRD analysis revealed that the polymer chains have been successfully intercalated into prGO layers. SEM has been used to verify the dispersion of the prGO particles in the matrix and to reveal the developed morphology of PP/EPR, in the presence of compatibilizers and functionalized prGO.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Graphene, a monolayer of sp2-hybridized carbon atoms bonded in the hexagonal lattice, has attracted enormous research interest in recent years [1]. As a typical two-dimensional material, graphene is the building block for other carbon structures such as fullerene, carbon nanotube (CNT) and graphite [2]. Because of its excellent properties, graphene is being explored for various applications in the fields of electronics, aerospace, automobile, defense industries, optics and sensors etc. [1]. Many attempts have been made to use graphene as inorganic naanoscale filler to enhance the electrical, physical, mechanical and barrier properties of polymer nanocomposites (PNCs) [3, 4].
This interest is directly related to the degree of dispersion of the graphene in the polymer matrix [5]. Other factors also play a major role in the properties of nanocomposites such as nanoparticle-matrix interaction and particle-particle interaction. The attraction forces between particles, due to the van der Waals and electronic forces affect the particle-particle interaction and deteriorate the nanocomposites performances. The improved mechanical properties can be achieved through improved interface between the particle and the matrix. The quality of interface and the strength of the adhesion at the interface determine load transfer between the matrix and the nanofillers [6].
To date, the mixing of graphene and functionalized graphene with polymers covers most of the published works, while direct modification of graphene with polymers is a somewhat less explored approach. However, in many cases, to achieve stable dispersions of graphene and adequate control of the microstructure of the nanocomposites, non-covalent or covalent functionalization of graphene may be necessary [7,8,9].
Recently, graphene oxide (GO) and its derivatives have emerged as one of the promising candidate to produce new PNCs because of their remarkable properties and abundance of its precursor; graphite [7] GO is generally produced by the treatment of graphite using strong minerals and oxidizing agents. This is typically achieved via treatment with KMnO4 and H2SO4, as in the Hummers method or its modified derivatives, or KClO3 and HNO3 as in the Staudenmaier or Brodie methods [10,11,12,13,14]. These reactions achieve similar levels of oxidation (C:O ratios of approximately 2:1) which ultimately disrupts the delocalized electronic structure of graphite and imparts a variety of oxygen-based chemical functionalities to the surface.
Compared to pristine graphene, GO is heavily oxygenated and its basal plane carbon atoms are decorated with epoxide and hydroxyl groups and its edge atoms with carbonyl and carboxyl groups. The introduction of oxygen-containing functional groups, results in an increase in the interlayer spacing from 3.4 Å of original graphite to more than 6 Å for GO depending on the water penetrated into the interlayer space of the platelets [15]. Hence, GO is highly hydrophilic and the presence of these functional groups reduces interplanar forces, which can improve the interfacial interaction between GO and some polymers and thus the dispersion state of GO in polymer matrices [7]. From the structure model of GO, proposed by Lerf et al. [16]. It is clear that GO has numerous reactive groups, making it potentially functionalized covalently with a great number of modifying reagents. Non-covalent interactions of GO primarily involve van der Waals interaction and π–π stacking.
However, one of the challenges remains the homogeneous dispersion of GO particles into the polymer matrix, which is influenced by the interfacial interactions between the GO nanosheets and the polymer matrix. Also, the ultimate properties of the nanocomposites are critically dependent upon the processing methods and the processing conditions. GO/PNCs can be processed using three synthesis routes: solution blending, in situ polymerization and melt mixing. Solution mixing is the most straightforward in which the polymer is soluble. The GO, for instance, is first swollen and dispersed in solvent before mixing it with the polymer solution. Eventually, on removal of the solvent, a multilayer structure is formed as the sheets reassemble trapping the polymer chains. However, solvent removal is a critical issue. Also, because of its functional groups, GO can be directly mixed with water-soluble polymer [17,18,19,20,21,22,23,24,25].
Dramatic improvement of solubility and interaction of nanofiller with polymer can be obtained by chemical functionalization of GO (f-GO) with isocyanate, amine or polymer grafted GO [26]. In situ polymerization is another efficient route for the preparation of homogeneously dispersed GO or its derivatives in a polymer matrix without a prior exfoliation solution with an initiator. In situ polymerization technique allows possible the covalent bonding between functionalized sheets and polymer matrix via various chemical reactions. However, this method requires monomer units and a large amount for polymerization [26,27,28,29,30,31]. Melt mixing is a typical standard approach for synthesizing thermoplastic PNCs. No solvent is required in this method and, GO or its derivatives are mixed mechanically at high temperatures and strong shear forces using conventional methods, like extrusion and injection molding. Several GO/PNCs have been prepared using this method, such as polystyrene (PS), polyethylene (PE) and polypropylene [32,33,34,35,36].
Thermoplastic elastomers (TPE) represent an important class of polymeric materials that combine the properties of thermo-plasticity during processing and rubber-like behavior in service. Among TPEs based on polymer blends, the most common are those prepared from polypropylene blends. A variety of polypropylene blends with ethylene-octene copolymer (POE), ethylene-propylene-diene (EPDM), ethylene-propylene copolymer (EPR), styrene block copolymer (SBS), have been commercialized for automotive applications such as bumpers, dashboards etc. These blends however, possess lower stiffness, which may be a serious shortcoming in certain applications. Incorporation of nanomaterials has attracted attentions due to the synergistic properties combining the two complementary technologies of thermoplastic elastomers and nanocomposites [37, 38].
In the present work structure and properties of the ternary phase PP containing EPR and partially reduced graphene oxide (prGO) are reported. The investigation is carried out on the PP/EPR mixture in the proportion of 85/15. The prGO nanoparticles were intercalated with dodecylamine (DDA) and functionalized according to the covalent grafting technique using two commercial organosilanes (Z6020 and A1100) and then, introduced into the polymer matrix at mass rates of 0.5, 1, 1.5 and 2 wt%. Three types of compatibilizing agents were used during this work in order to promote the interaction between the different phases involved, namely: diethyl maleate functionalized EPR (EPR-g-DEM), glycidylmethacrylate functionalized EPR (EPR-g-GMA) and maleic anhydride functionalized EPR (EPR-g-MAH). All Nanocomposites were prepared as a two-step compounding process. The mechanical properties (tensile strength and impact resistance) of the various materials were studied. The thermal stability of the nanocomposites was studied by thermogravimetric analysis (TGA). The morphology and crystalline structure of the prepared materials were analyzed by X-ray diffraction (XRD) and scanning electron microscopy (SEM).
Experimental
Materials and characterization
Graphite powder particles (20 μm) were purchased from Sigma-Aldrich. The chemicals used for the synthesis in graphene oxide (GO) such as sulfuric acid (H2SO4, 30%), hydrogen peroxide (H2O2, 30%), chlorhydric acid (HCl, 38%), sodium nitrate (NaNO3, 99%), sodium borohydride (NaBH4, 98.5%) and iodine (I2, 99%) were obtained from Sigma-Aldrich and used without further purification. Potassium permanganate (KMnO4, 99.5%), was purchased from Fisher and used as received. The intercalated material used in this work was dodecylamine (DDA, 99%) supplied by Sigma-Aldrich. The silanes used are N-(β-aminoethyl)-γ-aminopropyltrimethoxysilane (Z6020, > 98%) and γ-aminopropyltriethoxysilane (A1100, > 99%) were generously donated by Dow Corning Corp. (USA). The polypropylene (ISPLEN PP040 G1E) used as matrix is an isotactic homopolymer obtained from Repsol Quimica S.A. (Spain). The EPR rubber is a commercial grade product (Exxelor PE805) from Exxon chemical co (USA). The ethylene/propylene ratio of this material was 75/25. The compatibilizers used in this work were synthesized using the dicumyl peroxide (DCP, 98%) as the free initiator, diethyl maleate (DEM, 97%), glycidymethacrylate (GMA, 97%) and maleic anhydride (99%) as monomers. All these reagents were procured from Sigma-Aldrich and were used without further purification.
Preparation of graphene oxide (GO)
The conventional experimental Hummers method was used for the synthesis of graphene oxide (GO) [12]. 5 g of graphite and 2.5 g of fine sodium nitrate (NaNO3) were mixed with 115 ml of concentrated sulfuric acid (H2SO4) in a round-bottom flask and stirred continuously in an ice bath for 30 min. 15 g of potassium permanganate (KMnO4) was slowly added to the solution within 1 h to keep the mixture temperature not exceeding 2 °C; after being stirred for 3 h, the solution temperature was slowly added to the reaction vessel while keeping the temperature less than 98 °C. The diluted suspension was maintained at this temperature for 15 min. The suspension was further diluted to approximately 340 ml with warm distilled water and treated with 25 ml of hydrogen peroxide (H2O2) to reduce the residual KMnO4 and MnO2 to colorless soluble manganese sulfate. The mixture was filtered using the Whatman Anodisc membrane. The filtered cake was then, washed successively with chlorhydric acid (HCl, 5%V/V) in order to eliminate SO4 2− ions and with distilled water until the chloride test with silver nitrate (AgNO3) became negative. The bulk of the water was removed by oven drying at 65 °C for 36 h.
Preparation of partially reduced graphene oxide (prGO)
GO was dispersed in distilled water to get a 1 g/L colloidal solution. The pH of this solution was adjusted to 9 ~ 10 by 5 wt% sodium carbonate solution. Then, 1 g of NaBH4 was added into 100 ml GO dispersion under magnetic stirring at 80 °C for 2 h. Afterwards, 250 mg of I2 dispersed in 15 ml THF was added into the above suspension and stirred for 24 h. The obtained product was separated by filtration and sequentially washed with 1 M HCl, 1 M NaOH and large amounts of water several times to remove most residual ions The final product was dried under vacuum at 100 °C for 12 h then, grinded in fine powder and kept in a sealed container. The partially reduced GO was denoted as prGO.
Intercalation of prGO with dodecylamine
5 g of prGO was dispersed and swelled in 500 ml of distilled water. Then, 7 g of DDA, dissolved in 120 ml ethanol, were slowly added to the slurry over 3 h under vigorous stirring. The reaction continued for 72 h at 60 °C. Intercalated prGO was collected by filtration at ambient temperature, washed with 1:2 warm water/ethanol mixtures six times to eliminate the excess of surfactant. The obtained product was dried under vacuum at 80 °C for 24 h, then ground, pulverized to a fine powder and kept in a sealed container. The intercalated prGO was designated as DDA-prGO.
Silylation of DDA-prGO with silanes
The silylation process of graphene oxide with silanes was carried out in a solution of ethanol/water at a ratio of 75:25 (v/v). About 10 wt% of each silane were introduced in 500 ml of the mixture and stirred vigorously with a magnetic stirrer for 3 h at 60 °C. Then, 2 g of DDA-prGO were added to the above-mentioned solution and the grafting reaction was achieved under vigorous stirring during 5 h at 80 °C. The product was separated by filtration and sequentially washed with ethanol then with acetone repeatedly in order to remove any residual silane. The modified product was dried in a vacuum oven at 105 °C for 24 h, then pulverized in an agate mortar to pass a 200 mesh sieve and stored for further use. The functionalized samples prepared from the silanes were denoted as Z6020-prGO and A1100-prGO.
Preparation of compatibilizers
The grafting reaction was carried out in the molten state using a 50 ml Brabender plastograph PL2100. Typically, 2 wt% of monomers and 0.1 wt% of DCP were introduced to the EPR matrix in the preheated chamber. The reaction was conducted at 190 °C and mechanically mixed at 60 rpm for 10 min. The product was pelletized and purified in xylene for 24 h to remove any unreacted monomer. Finally, the product was dried in a vacuum oven at 60 °C to constant weight and kept firmly away from the humidity in plastic bags. The functionalized EPR prepared from DEM, GMA and MAH were denoted as EPR-g-DEM, EPR-g-GMA and EPR-g-MAH, respectively.
Preparation of nanocomposites
All nanocomposites were prepared as a two-step compounding process. First, a masterbatchs were prepared by dispersion of the prGO or functionalized prGO within the compatibilizers dissolved in p-xylene at 120 °C. The resulting mixtures were refluxed under magnetic stirring for 24 h. Afterwards, the solvent was evaporated and the samples were dried. The masterbatchs were incorporated into PP/EPR matrix in an 85/15 ratio by melt blending using Brabender PL2100 mixer for 8 min at 200 °C and 40 rpm. After pelletizing, the nanocomposite granules were compression molded using a hydraulic press at 200 °C and 200 Kg/cm2 for 6 min. The weight fractions of the nanofillers in the matrix were 0, 0.5, 1, 1.5, and 2 wt%. The compatibilizer’s content was fixed at 10 wt%. Table 1 gives the compositions and the sample codes of the prepared nanocomposites. There are different types of nanocomposites with various types of nanofillers and compatibilizers. I01, I02 and I03 designate the non-compatibilized nanocomposites samples containing pristine prGO, Z6020-prGO and A1100-prGO, respectively. The neat PP/EPR (i.e. without prGO) sample is called I010. The nanocomposites samples prepared using 0.5, 1, 1.5 and 2 wt% are designated (i.e pristine prGO) I010.5, I011, I011.5 and I012, respectively. The neat compatibilized blends: PP/EPR/EPR-g-DEM, PP/EPR/EPR-g-GMA and PP/EPR/EPR-g-MAH are designated I100, I200 and I300. I11 and I12 stand for compatibilized nanocomposites samples (i.e. PP/EPR/EPR-g-DEM) containing Z6020-prGO and A1100-prGO, respectively. The nanocomposites generated from I11 and I12 (i.e. with 1 wt% of modified prGO) are denoted I111 and I121.
ATR-FTIR spectroscopy
About 50 mg of the powders were compacted into thin pellets using a hydraulic press under a force of 5 tons maintained for about 4 min. Samples were analyzed by signal averaging 16 scans at a resolution of 4 cm−1 using a JASCO-IR spectrometer. FTIR analysis was also performed to confirm the grafting reactions of EPR. Thin films were obtained from the samples by compression molding between two Teflon sheets using a compression machine at 200 °C and 200 kg/cm2. The spectra were recorded on a Shimatzu spectrometer in the region of 4000–400 cm−1 at resolution of 4 cm−1 with 32 coadded scans.
Thermogravimetric analysis (TGA)
TGA measurements studies of both nanofillers and their nanocomposites were performed using a Setaram SETSYS TG-DTA thermal analyser under nitrogen atmosphere at a flow rate of 20 cm3/min. About 10 to 20 mg of each sample was heated from 25 to 600 °C with a heating rate of 10 °C/min.
X-ray diffraction (XRD)
XRD analysis was performed at room temperature with copper radiation using a Phillips X’Pert diffractometer at the step size of 0.02° (2θ) and the rate of 2.4° (2θ) per minute. The X-ray beam was a CuKα radiation (λ = 1.54 Å) using a 35 kV voltage generator and a 55 mA current. The basal interlayer of the samples was estimated from the position of the d002 plane peak in the X-ray diffraction intensity profile using Bragg’s law [39].
Mechanical testing
Tensile properties were performed using an Instron MTC 500 at a crosshead speed of 20 mm/min. From the stress-strain curves; the tensile strength, Young modulus and elongation at break were determined. Tensile properties were performed according to ASTM D 638 specifications. Impact testing was done using a Ceast impact pendulum tester at impact energy of 7.5 kJ according to ASTM D 256–88. The hammer velocity was set at 2.88 m/s while the weight of the hammer was 1.8 Kg. All measurements were made in five replicants and the values were averaged.
Scanning electron microscopy (SEM)
The morphology of the nanocomposites was observed with a Quanta 200 Scanning Electron Microscope at an acceleration voltage of 10 kV. Test specimens, produced by compression molding at 200 °C and 180 Kg/cm2, were frozen by immersion in liquid nitrogen to obtain brittle fracture. Gold sputter coating was not required for the preparation of the samples.
Results and discussion
ATR-FTIR analysis of the prGO and its derivatives
The ATR-FTIR spectra of graphite and its corresponding GO and prGO are shown in Figs. 1 and 2. In Fig. 2a, the characteristic band at 1580 cm−1 was due to the graphitic structure [40]. After oxidation (Fig. 2b), the presence of characteristic bands of OH and COOH stretching at 3692, 3625 and 3428 cm−1 and C = O stretching at 1725 cm−1 are evident in oxidized sample. Other bands appeared at 1624 cm−1, 1390 cm−1, 1228 cm−1 and 1059 cm−1, which corresponds to quinone groups, O-H bending, epoxy groups and C-O stretching, respectively. These results confirm the efficiency of oxidation. In the spectrum of partially reduced graphene oxide (Fig. 2c), the disappearance of the C = O stretching around 1725 cm−1, C-O stretching at 1228 cm−1 and the higher intensity of the O-H band confirm the conversion of all C-O-C groups and COOH groups into C-OH groups. Figure 3 shows the FTIR spectra of intercalated and silanized prGO in comparison with prGO. The spectrum of DDA-prGO (Fig. 3b) exhibits two strong bands at 2925 cm−1 and 2849 cm−1, which are attributed to -CH asymmetric and symmetric stretching of CH2 groups in DDA respectively. The observed band at 1525 cm−1 can be ascribed to the symmetric or the asymmetric deformation band of NH3 +. This indicates that the DDA molecules interact onto the surface through hydrogen bonding between the amino groups and the hydroxyl groups of prGO (−CH2-O-H…H2N-R). Similar results have been reported by Y. Matsuo et al. [41] and I.V. Shernyshova et al. [42] for alkylamines intercalated graphite oxides and alkylamines intercalated silicates, respectively. For the silanized samples (Fig. 3c and d), the higher intensity at 2930 cm−1 and 2854 cm−1 comes from CH2 and the appearance of the Si-O stretching at 1080 cm−1 confirm the success of silanization.

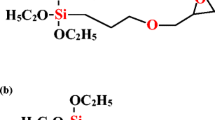
Chemical formulas of intercalate and silane coupling agents a DDA b Z6020 and c A1100

ATR-FTIR spectra of a pristine graphite, b graphite oxide (GO) and c partially reduced graphene oxide (prGO)

ATR-FTIR spectra of a prGO, b DDA-prGO, c Z6020-prGO and d A1100-prGO
XRD analysis of the prGO and its derivatives
XRD patterns of prGO, DDA-prGO, Z6020-prGO and A1100-prGO as well as that of pristine graphite and GO are shown in Fig. 4. The pattern of the pristine graphite (Fig. 4a) shows a very strong (002) peak at 2θ = 26.3°, for a typical graphitic structure with a layer spacing of 0.34 nm. The intensity of this peak sharply decreases for GO (Fig. 4b), and a new diffraction peak appears at about 2θ = 16°. The d001 interlayer distance calculated from Bragg’s law, is 0.56 nm, which is due to the formation of oxygen-containing functional groups, such as hydroxyl, epoxy, carboxyl and intercalated water [43]. Figure 4c shows a diffractogram of a slight displacement of 2θ angle to higher values, after reduction. The peak appears at2θ = 17.4°, which corresponds to a layer spacing of 0.5 nm. This value is higher than that of the interlayer spacing of pristine graphite due to some oxygenated functional groups trapped in interlayer space. This means that GO is partially reduced. After intercalation and silanization processes (Fig. 4d-f), the diffractograms shows a displacement of the 2θ angle to lower values. The peak positions for all the samples is at 2θ = 13° value, which corresponds to a new layer spacing of 0.687 nm. This increase of layer spacing may be attributed to the intercalation of organic and water molecules within the graphenic layers. However, silanization processes has no apparent effect on the interlayer spacing, this suggests that the silanization is non-destructive to the structure of graphitic crystallite.

XRD patterns of a pristine graphite, b GO c prGO, d DDA-prGO, e Z6020-prGO and f A1100-prGO
TGA analysis of the prGO and its derivatives
Thermal stability of pristine graphite, GO, and prGO and its derivatives was analyzed using TGA (Fig. 5). TGA curve of pristine graphite shows a high thermal stability (Fig. 5a). No weight loss was observed in the temperature range investigated. However, the thermal degradation profile for GO sample (Fig. 5b) exhibits two weight loss steps at 80 °C and at 250 °C. The first step is due to the evaporation of adsorbed water and the second one is due to the decomposition of unstable oxygen-containing functionalities generating carbon dioxide and water [44]. The thermogram of prGO (Fig. 5c) exhibits also two weight loss steps. The first one occurring at 105 °C is due to the release of physisorbed water of the sample. The second one appearing at an onset temperature of 260 °C (vs 250 °C onset obtained for GO), is attributed to the decomposition of residual stable oxygen after partial reduction process. The TGA curve obtained for DDA-prGO sample (Fig. 5d) shows a weight loss at 120 °C due to the release or transformation of some-oxygen-containing groups either by reaction with DDA. The weight loss observed at 360 °C is attributed to the decomposition of the intercalant. The TGA curves obtained for silylated materials show three steps of weight loss. The weight loss at about 100 °C is primarily due to the presence of water within sample. The minor weight loss observed at about 250 °C is presumably due to the decomposition of residual oxygenated functional groups that did not react with silane molecules. The last weight loss observed at 380 °C is assigned to the decomposition of silane molecules.

TGA curves of a Pristine graphite b GO, c prGO, d DDB-prGO, e Z6020-prGO and f A1100-prGO, respectively
FTIR analysis of the compatibilizers
The grafting of EPR is confirmed by FTIR spectroscopy. The FTIR spectra of EPR-g-DEM, EPR-g-GMA and EPR-g-MAH are shown in Fig. 6. The reference spectrum (Fig. 6a) does not show any band in the 2000–1500 cm−1 region. In Fig. 6b, the FTIR spectrum of EPR-g-DEM presents a strong characteristic band at 1740 cm−1 (C = O symmetric stretching band) which indicates the presence of carbonyl groups. The spectrum of EPR-g-GMA (Fig. 6c) shows one band at 1730 cm−1characteristic of the carboxyl groups of GMA. The spectrum of EPR-g-MAH (Fig. 6d) shows three bands at 1850 cm−1, 1785 cm−1 and 1712 cm−1. According to the literature [45], the first two bands are generally attributed to carbonyl (C = O) groups associated with anhydride maleic and the last one is attributed to the vibration of the carboxyl (C = O) groups of an acid function. This result is justified by the opening of the maleic anhydride ring because of its great susceptibility to hydrolysis [46, 47].

FTIR spectra in the range 2000–1500 cm−1 of a EPR, b EPR-g-DEM, c EPR-g-GMA and d EPR-g-MAH
Mechanical properties of the blends and nanocomposites
The tensile and Izod impact strength of PP and the different blends are summarized in supplementary Table S1 (Online Resource). in the supporting information. As expected it is observed that the incorporation of EPR in PP improves ductility of the blend. This is indicated by the elongation at break and the impact strength of PP, which increased by 150% and 50%, respectively. Similar improvements in ductility with the incorporation of elastomers have been also reported by previous researchers [48, 49]. However, the tensile strength and Young’s modulus decreased by about 31% and 19%, respectively as compared to the neat PP. Those results are consistent also with previous studies [50,51,52,53]. A decrease in tensile strength and Young’s modulus can be explained by the substitution of PP by EPR, which is a low modulus material, and by the morphological changes due to EPR phase [48, 49, 54, 55].
The improvement in elongation at break and impact strength can be explained either by the influence of functionalized rubber on the deformation mechanisms in the blend, or by the alteration of blend microstructures due to the presence of a third phase [50]. Similar improvements in ductility of PP/EPR with the incorporation of PEP and EPR-g-MAH have been reported by previous researches [56]. It is also interesting to note that the elongation at break and impact strength of the PP/EPR increased by about 30% and 42% respectively, in the presence of EPR-g-DEM, as compared to the non-compatibilized PP/EPR blend.
Tensile strength and Young’s modulus of all compositions of the nanocomposites were reported in Figs. 7 and 8. It can be seen that the addition of nanofiller particles led to a substantial improvement in stiffness. The tensile strength and Young’s modulus of the nanocomposites increased with the increasing prGO content from 0 to 2 wt%. With 2 wt% nanofiller, tensile strength and Young’s modulus improved by about 40% and 14%, respectively, as compared to the unfilled PP/EPR blend. This increase in tensile strength and Young’s modulus of all nanocomposites indicates an increase in the rigidity of polymer matrix related to the restriction of the mobility in polymer chains due to the presence of filler nanoparticles.

Effect of compatibilizers and prGO content on tensile strength of PP/EPR nanocomposites

Effect of prGO content and compatibilizers on Young’s modulus of PP/EPR nanocomposites
Addition of compatibilizers resulted in an improvement of the tensile strength and Young’s modulus. It is important to note that the EPR-g-DEM compatibilized nanocomposites showed higher elongation at break and impact strength, as compared to EPR-g-GMA and EPR-g-MAH compatibilized nanocomposites.
Supplementary Figs. S1 and S2 (Online Resource) show the influence of the prGO content and compatibilizers on elongation at break and Izod impact strength of the nanocomposites. It can be seen that elongation at break and impact strength decreased as the nanofillers content increased from 0 to 2 wt%. This decrease may be due to the poor adhesion or the absence of bonding at the interface between the matrix and the nanofiller. The poor interfacial adhesion causes premature failure as a result of the usual crack opening mechanism. Addition of compatibilizers to the nanocomposite systems caused a considerable reduction of elongation at break and impact strength. When EPR-g-DEM was used as a compatibilizer, the elongation at break and impact strength of nanocomposites decreased by 72% and 69%, respectively. This decrease indicates that the nanocomposites become brittle with increasing prGO content owing to the stress concentration effect of nanofiller.
Figures 9 and 10 show the influence of the functionalization of prGO on tensile strength and Young’s modulus of the non-compatibilized nanocomposites and compatibilized containing 1 wt% of prGO, respectively. As observed from the above Figures, silane functionalization leads to an increase in tensile strength and Young’s modulus by improving adhesion between prGO and polymer matrix. Higher values of the tensile strength and Young’s modulus were observed for the nanocomposites containing prGO functionalized with Z6020 and compatibilized with EPR-g-DEM. In this system, the tensile strength and Young’s modulus increases by about 31% and 23%, respectively compared to the nanocomposites without silane coupling agent. The amino groups anchored on Z6020-prGO and A1100-prGO surface may interact with the functional groups of compatibilizers, resulting in improved compatibility of the prGO with the polymer matrix. However, when EPR-g-GMA is used as a compatibilizer, the tensile strength and Young’s modulus of nanocomposites increase slightly. There may be no effective interactions between the GMA functionalized EPR and the functional groups on the surface of the nanofiller particles.

Tensile strength of non-compatibilized and compatibilized PP/EPR nanocomposites containing 1 wt% untreated and treated prGO

Young’s modulus of non-compatibilized and compatibilized PP/EPR nanocomposites containing 1 wt% untreated and treated prGO
The elongation at break and the impact strength of the nanocomposites containing 1 wt% of prGO are shown in supplementary Figs. S3 and S4 (Online Resource). The results showed that the incorporation of functionalized prGO has substantially reduced the elongation at break, as well as the impact strength of the nanocomposites. The most important drop in the elongation at break and impact strength is observed with functionalized prGO-based nanocomposites. This reduction becomes more pronounced with the incorporation of compatibilizers. The results revealed that the elongation at break and impact strength of PP/EPR/EPR-g-DEM/Z6020-prGO decreases by about 55% and 26%, respectively as compared to the same nanocomposites without coupling agent and about 84% and 67% respectively, compared to the neat matrix. Consequently, the decrease of these properties can be attributed to the strong improvement of the interfacial region, reflecting the strong interfacial adhesion between the different components.
Thermal behaviour of the blends and nanocomposites
Thermal stability of PP/EPR blend and PP/EPR/prGO nanocomposites were studied by TGA as shown in supplementary Fig. S5 (Online Resource). From the observation, it is clear that the thermal stability of the base polymer matrix increased with the incorporation of the nanofiller. During the thermal decomposition, the TGA curves displayed a single stage decomposition process for all samples. It can be seen that the onset decomposition (Tonset) and fasted degradation (Tmax) of different nanocomposites are higher that the neat PP/EPR matrix. It is also noted that the Tonset and Tmax remained almost unchanged for nanocomposites with the prGO content. The addition of the nanofiller which is in a lamellar form leads to an important improvement of the thermal stability of PP/EPR blend. The Tonset and Tmax increased from 360 °C to 390 °C and from 440 °C to 460 °C respectively, which about 30 °C and 20 °C, respectively higher than the nanocomposite with 0.5 wt% prGO additions than that for neat matrix. Further additions of prGO up to 2 wt% led to further improvement of the thermal stability of nanocomposites. It was noticed that the Tonset and Tmax reached 400 °C and 475 °C respectively corresponding to an increase by 40 °C and 30 °C respectively, compared to the neat matrix.
Figure 11 shows the thermogravimetric analysis of the blends and nanocomposites with and without compatibilizers and coupling agents. The incorporation of the compatibilizer does not show any effect on the decomposition process in comparison to the neat matrix. The process of degradation in PP/EPR/EPR-g-DEM/prGO and in all other materials containing silane coupling agents was similar. The Tonset and Tmax shifted to 403 °C and 480 °C, an increase of 43 °C and 35 °C respectively, compared to the PP/EPR and PP/EPR/EPR-g-DEM blends. It seems that the filler can act as a barrier to hinder the permeability of volatile decomposition products out of the material [57]. This effect is accentuated in the presence of the compatibilizers and silane coupling agents. This may be attributed to a well dispersion and distribution the prGO particles in the polymer matrix. The increase in the thermal stability can also be attributed to the polymer/nanofiller interactions due to the compatibilizer and silane coupling agent’s effect. Numerous researchers have reported a similar improvement in the thermal stability for a large number of polymer nanocomposites based on graphene or its derivatives [58,59,60]. Several factors may contribute to achieve a high thermal performance of graphene/polymer nanocomposites such as complete exfoliation and homogeneous dispersion of graphene layers in polymer matrix and strong interaction of graphene layers with matrix.

TGA curves of PP/EPR/EPR-g-DEM/prGO nanocomposites containing 1 wt% untreated and treated prGO
X-ray diffraction of the blends and nanocomposites
The XRD patterns for neat PP, PP/EPR and PP/EPR/EPR-g-DEM blends are given in Fig. 12. PP shows six characteristic peaks at 2θ = 14.3° (0.62 nm), 16.9° (0.53 nm), 18.6° (0.48 nm), 21.3° (0.42 nm) 23.7° (0.38 nm), 25.45° (0.36 nm) [38, 39]. We notice that the diffraction peaks are slightly affected in their position and intensity by the incorporation of EPR and EPR-g-DEM, which would imply a modification of the cristallinity. Figure 13 shows the XRD diffraction patterns of neat PP/EPR blend and PP/EPR/prGO nanocomposites containing 1 wt% of pristine and functionalized prGO with Z6020. The XRD patterns of the non-compatibilized nanocomposites have all the peaks observed in both prGO and PP/EPR matrix polymer. However, the XRD profiles of nanocomposites show a decrease in intensity for all peaks observed for PP/EPR blend. In addition, the peak of prGO shifted from 2θ = 17.4° to lower angles of about 2θ = 16° and 2θ = 14.77° for PP/EPR/prGO and PP/EPR/Z6020-prGO respectively, as compared to that of pristine prGO. This implies that the interlayer spaces between sheets were increased from 0.5 nm to 0.56 nm and 0.6 nm. The shift towards lower angles, especially in the case of PP/EPR/Z6020-prGO clearly indicates a basal spacing expansion and an intercalation of PP/EPR chains into the layers without the presence of compatibilizer. This may be due to the alkylamine pre-intercalated prGO which had successfully reduced the incompatibility of the functional groups on the surface of the prGO nanoparticles and the non-polar polymer matrix. This XRD behaviour is similar to that obtained in polymer/clay nanocomposites by others [61, 62].

XRD patterns for PP, PP/EPR and PP/EPR/EPR-g-DEM blends

XRD patterns for neat PP/EPR blend, PP/EPR/prGO (1 wt%) and PP/EPR/Z6020-prGO (1 wt%) nanocomposites
The XRD patterns for compatibilized nanocomposites with EPR-g-DEM in comparison with XRD pattern of PP/EPR/EPR-g-DEM (Fig. 14), clearly show a decrease in intensity of all peaks observed for the unfilled system. This would imply a modification of the polymer structure. As indicated in the XRD diffractograms, the peak of prGO shifted from 2θ = 17.4° to lower angles of about 2θ = 11.23° and 2θ = 10.54° for PP/EPR/EPR-g-DEM/prGO and PP/EPR/EPR-g-DEM/Z6020-prGO respectively. This corresponds to an interlayer spacing of 0.8 nm and 0.85 nm, respectively; indicating that incorporation of EPR-g-DEM would contributes to a better intercalation.

XRD patterns for neat PP/EPR/EPR-g-DEM, PP/EPR/EPR-g-DEM/prGO (1 wt%) and PP/EPR/EPR-g-DEM/Z6020-prGO (1 wt%) nanocomposites
SEM micrograph of the blends and nanocomposites
The morphologies of blends are shown in Fig. 15. It is obvious that the particle size of the dispersed EPR phase in PP is affected by the type of compatibilizer. Indeed the SEM micrographs of PP/EPR blend (Fig. 15a) shows a nodular morphology, where the elastomer phase is dispersed in a spherical form in PP. Debonding of EPR particles from the matrix are also observed at the fractured surface.

SEM micrographs of a PP/EPR, b PP/EPR/EPR-g-DEM, c PP/EPR/EPR-g-GMA and d PP/EPR/EPR-g-MAH blends
A fine and uniform dispersion of EPR particles in the PP phase is observed (Fig. 15b). The decrease in the particles size of the dispersed EPR phase is due to the reduction of the interfacial tension between the dispersed EPR phase and PP matrix. On the other hand, the number of particles is higher than that obtained from the original blend. In the case of PP/EPR blend with EPR-g-GMA and EPR-g-MAH (Fig. 15c and d) the size and the number of the dispersed elastomer particles are higher. Hence, the presence of these compatibilizers reduces the interfacial tension and suppresses the coalescence of EPR particles.
The morphology of the nanocomposites characterized by the SEM micrographs is presented in Fig. 16. We can see that the micrograph of PP/EPR/Z6020-prGO (Fig. 16a) shows that the Z6020-prGO particles are not dispersed in a homogeneous manner throughout the matrix. However, the presence of the compatibilizers has in fact promoted a better dispersion of the functionalized prGO in the matrix.

SEM micrographs of a PP/EPR/Z6020-prGO, b PP/EPR/EPR-g-DEM/ Z6020-prGO, c PP/EPR/EPR-g-GMA/Z6020-prGO and d PP/EPR/EPR-g-MAH/ Z6020-prGO nanocomposites
Figure 16b-d illustrates the SEM micrographs of the compatibilized nanocomposites. It is shown that the use of the compatibilizers has significantly promoted a better dispersion of the functionalized prGO in the matrix. On the other hand the nanocomposite prepared using EPR-g-DEM shows a more pronounced reduction of the size domains of the EPR. This corroborates the good tensile properties that were obtained with the latter system.
Conclusions
The nanocomposites of polypropylene/ethylene-propylene rubber (PP/EPR) with different amounts of functionalized partially reduced graphene oxide and three types of compatibilizers were investigated to determine their influence on the mechanical, thermal and morphological properties. The nanocomposites containing compatibilizers and functionalized prGO showed a significant improvement of the mechanical properties compared to the nanocomposites containing only pristine prGO without compatibilizers. This is due to the enhancement of interfacial adhesion and compatibility between polymer matrix and functionalized prGO. The incorporation of pristine or functionalized prGO as well as compatibilizers into the PP/EPR matrix, results in a remarkable improvement in the thermal stability of nanocomposites and also the successful intercalation of polymer chains into prGO interlayer spaces. SEM micrographs reveal that PP/EPR and PP/EPR/compatibilizers have a nodular microstructure. The presence of compatibilizers in PP/EPR/prGO nanocomposites promotes a better dispersion of functionalized prGO particles in the polymer matrix.
References
An X, Simmons T, Shah R, Wolfe C, Lewis Kim M, Washington M, Nayak Saroj K, Talapatra S, Kar S (2010) Stable aqueous dispersions of noncovalently functionalized graphene from graphite and their multifunctional high-performance applications. Nano Lett 10(1):4295–4301
Stoller MD, Park S, Zhu Y, Ruoff RS (2008) Graphene-based ultracapacitors. Nano Lett 8(10):3498–3502
Zaman I, Kuan HC, Dai J, Kawashima N, Michelmore A, Dong S, Luong L, Ma J (2012) From nanotubes and silicate layers to graphene platelets for polymer nanocomposites. Nano 4(15):4578–4586
Steurer P, Wissert R, Thamann R, Mülhaupt R (2009) Functionalized graphenes and thermoplastic nanocomposites based upon expanded graphite oxide. Macromol Rapid Commun 30(4–5):316–329
Hussain F, Hojjati M, Okamoto M, Gorga RE (2006) Polymer-matrix nanocomposites, processing, Manufacturing and Application: An Overview. J Compos Mater 40(17):1511–1575
Bhattacharya M (2016) Polymer nanocomposites - a comparison between carbon nanotubes, graphene and clay as nanofillers. Materials 9(4):262–297
Galpaya D, Wang M, Liu M, Motta N, Waclawik E, Ya C (2012) Recent advances in fabrication and characterization of graphene-polymer nanocomposites. Graphene 1:30–49
Xu Y, Liu Z, Zhang X, Wang Y, Tian J, Huang Y, Ma Y, Zhang X, Chen Y (2009) A graphene hybrid material covalently functionalized with porphyrin: synthesis and optical limiting property. Adv Mater 21(12):1275–1279
Yang H, Li F, Shan C, Han D, Zhang Q, Niu L, Ivaska A (2009) Covalent functionalization of chemically converted graphene sheets via silane and its reinforcement. J Mater Chem 19(26):4632–4638
Brodie BC (1859) On the atomic weight of graphite. Philos Trans R Soc 149:249–259
Staudenmaier L (1898) Verfahren zur darstellung der graphitsaure. Ber Dtsch Chem Ges 31(2):1481–1487
Hummers WS, Offeman RE (1958) Preparation of graphitic oxide. J Am Soc 80(6):1339–1339
Paulchamy B, Arthi G, Lignesh BD (2015) A simple approach to stepwise synthesis of graphene oxide nanomaterial. J Nanomed Nanotechnol. doi:10.4172/2157-7439.1000253
Shahriary L, Athawale AA (2014) Graphene oxide synthesized by using modified hummers approach. Int J Renew Energy Environmental Eng 2(1):58–63
Hontoria-Lucas C, López-Peinado A, López-González J, Rojas-Cervantes M, Martín-Aranda R (1995) Study of oxygen-containing groups in a series of graphite oxides: physical and chemical characterization. Carbon 33(11):1585–1592
Lerf A, He HY, Forster M, Klinovski J (1998) Structure of graphite oxide revisited. J Phys Chem B 102(23):4477–4482
Kuilla T, Bhadra S, Yao D, Kim NH, Bose S, Lee JH (2010) Recent advances in graphene based polymer composites. Prog Polym Sci 35(11):1350–1375
Mo ZL, Xie TT, Zhang JX, Zhao YX, Guo RB (2012) Synthesis and characterization of nanoGs-PPy/epoxy nanocomposites by in situ polymerization. Synth React Inorg, Met-Org, Nano-Met Chem 42(8):1172–1176
Bao C, Song L, Xing W, Yuan B, Wilkie CA, Huang J, Guo Y, Hu Y (2012) Preparation of graphene by pressurized oxidation and multiplex reduction and its polymer nanocomposites by masterbatch-based melt blending. J Mater Chem 22(13):6088–6096
Zhao X, Zhang Q, Chen D (2010) Enhanced mechanical properties of graphene-based poly(vinyl alcohol) composites. Macromolecules 43(5):2357–2363
Layek RK, Samanta S, Nandi AK (2012) The physical properties of sulfonated graphene/poly(vinyl alcohol) composites. Carbon 50(3):815–827
Jinhong Y, Huang X, Wu C, Jiang P (2011) Permittivity, thermal conductivity and thermal stability of poly(vinylidene fluoride)/graphene nanocomposites. IEEE Trans Dielectr Electr Insul 18(2):478–484
Chen Y, Qi Y, Tai Z, Yan X, Zhu F, Xue Q (2012) Preparation, mechanical properties and biocompatibility of graphene oxide/ultrahigh molecular weight polyethylene composites. Eur Polym J 48(6):1026–1033
Gonçalves G, Marques Paula AAP, Barros-Timmons A, Bdkin I, Singh Manoj K, Nazanin Emamic N, Grácioa J (2010) Graphene oxide modified with PMMA via ATRP as reinforcement filler. J Mater Chem 20(44):9927–9934
Kim H, Miura Y, Macosko CW (2010) Graphene/polyurethane nanocomposites for improved gas barrier and electrical conductivity. Chem Mater 22(11):3441–3450
Ren PG, Yan DX, Chen T, Zeng BQ, Li M (2011) Improved properties of highly oriented graphene/polymer nanocomposites. J Appl Polym Sci 121(6):3167–3174
Chatterjee S, Wang JW, Kuo WS, Chu BTT (2012) Mechanical reinforcement and thermal conductivity in expanded graphene nanoplatelets reinforced epoxy composites. Chem Phys Lett 531:6–10
Potts JR, Lee SH, Alam TM, An J, Stoller MD, Piner RD, Ruoff RS (2011) Thermomechanical properties of chemically modified graphene/poly(methyl methacrylate) composites made by in situ polymerization. Carbon 49:2615–2623
Zhang F, Peng X, Yan W, Peng Z, Shen Y (2011) Nonisothermal crystallization kinetics of in situ nylon 6/graphene composites by differential scanning calorimetry. J Polym Sci B Polym Phys 49(49):1381–1388
Wang X, Song L, Yang H, Xing W, Lu H (2011) In situ polymerization of graphene nanosheets and polyurethane with enhanced mechanical and thermal properties. J Mater Chem 21:4222–4227
Huang YF, Lin CW (2012) Facile synthesis and morphology control of graphene oxide/polyaniline nanocomposites via in-situ polymerization process. Polymer 53(13):2574–2582
Lian J, Wang Y, Huang Y, Maa Y, Liua Z, Caib J, Zhang C, Gao H, Chen Y (2009) Electromagnetic interference shielding of graphene/epoxy composites. Carbon 47(3):922–925
Kim H, Kobayashi S, Abdurrahim MA, Zhang MJ, Khusainova A, Hillmyer MA, Abdala AA, Macosko CW (2011) Graphene/polyethylene nanocomposites: effect of polyethylene functionalization and blending methods. Polymer 52(8):1837–1846
Sánchez-Valdes S, Zapata-Domínguez AG, Martinez-Colunga JG, Mendez-Nonell J, Ramos de Valle LF, Espinoza-Martinez AB, Morales-Cepeda A, Lozano-Ramirez T, Lafleur PG, Ramirez-Vargas E (2016) Influence of functionalized polypropylene on polypropylene/graphene oxide nanocomposite properties. Polym Compos. doi:10.1002/pc.24077
Kornmann X, Linderberg H, Bergund LA (2011) Synthesis of epoxy–clay nanocomposites: influence of the nature of the curing agent on structure. Polymer 42(4):4493–4499
Strumberger N, Gospocic A, Bratulic C (2005) Polymeric materials in automobiles. Promet Traffic Traffico 14(3):149–160
Mehta S, Mirabella FM, Rufener K, Bafna A (2004) Thermoplastic olefin/clay nanocomposites: morphology and mechanical properties. J Appl Polym Sci 92(2):928–936
Lim JW, Hassan A, Rahmat AR, Wahit MU (2006) Rubber-toughened polypropylene nanocomposite: effect of polyethylene octene copolymer on mechanical properties and phase morphology. Appl Polym 99(6):3441–3450
David IF, Shankland K, McCusker LB, Baerlocher C (2002) Structure determination from powder diffraction data. Oxford University Press Inc, New York,
Friedel RA, Carlson GL (1971) Infrared spectra of ground graphite. J Phys Chem 75(8):1149–1151
Matsuo Y, Miyabe T, Fukutsuka T, Sugie Y (2012) Preparation and characterization of alkylamine-intercalated graphite oxide. Carbon 45(5):1005–1012
Chernyshova IV, Rao KH, Vidyadhar A (2000) Mechanism of long-chain alkylamines on silicates. A spectroscopic study. 1. Quartz Langmuir 16 (21): 8071–8084
Tong X, Wang H, Wang G, Wan L, Ren Z, Bai J (2011) Controllable synthesis of graphene sheets with different numbers of layers and effect of the number of graphene layers on the specific capacity of anode material in lithium-ion batteries. J Solid State Chem 184(5):982–989
Herrera-Alonso M, Abdala AA, McAllister JM, Aksay IA, Prud’homme RK (2007) Intercalation and stitching of graphite oxide with diaminoalkanes. Langmuir 23(21):10644–10649
Bellamy LJ (1984) The infrared spectra of complexes molecules. Wiley, New York,
Doufnoune R, Haddaoui N, Riahi F (2006) Elaboration and characterization of an organic/inorganic hybrid material: effect of the interface on the mechanical en thermal behavior of PP/CaCO3 composites. Int J Polym Mater 55(10):815–834
Doufnoune R, Haddaoui N, Riahi F (2008) The effects of coupling agents on the mechanical, rheological and thermal properties of calcium carbonate-filled LDPE compatibilized with maleic anhydride-g-LDPE (part II). Int J Polym Mater 57(7):690–708
D'Orazio L, Mancarella C, Martuscelli E, Polato F (1991) Polypropylene/ethylene-co-blends influence of molecular structure and composition of EPR on melt rheology, morphology and impact properties of EPR injection molded samples. Polymer 32(7):1186–1194
Greco R, Mancarella C, Martuscelli E, Ragosta G, Jinghua Y (1987) Polyolefin blends, 2: effect of EPR composition on structure, morphology and mechanical properties of iPP/EPR alloys. Polymer 28(11):1929–1936
Premphet K, Horanont P (2000) Phase structure of ternary polypropylene/elastomer/filler composites: effect of elastomer polarity. Polymer 41(26):9283–9290
Premphet K, Horanont P (2000) Phase structure and property relationships in ternary polypropylene/elastomer/filler composites: effect of elastomer polarity. J Appl Polym Sci 76(13):1929–1939
Premphet K, Horanont P (1999) Influence of stearic acid treatment of filler particles on the structure and properties of ternary-phase polypropylene composites. J Appl Polym Sci 74(14):3445–3454
Frounchi M, Dadbin S, Salehpour Z, Noferesti M (2006) Gas barrier properties of PP/EPDM blend nanocomposites. J Membr Sci 282(1–2):142–148
Pukanszky B, Tudos E, Kallo A, Bodor G (1989) Multiple morphology in polypropylene/ethylene-propylene-diene terpolymer blends. Polymer 30(8):1407–1413
Zebarjad SM, Bagheri R, Lazzeri A (2001) Hybrid PP–EPR–GF composites, part 1- deformation mechanisms. Plast Rubber Compos 30(8):370–376
Mnif N (2008) Elaboration et caractérisation de mélanges complexes à base de polypropylène en vue de son écoconception et de son recyclage dans les véhicules hors d’usage, Ph. D thesis, Institut National des Sciences Appliquées de Lyon, France
Yoo BM, Shin HJ, Yoon HW, Park HB (2014) Graphene and graphene oxide and their uses in barrier polymers. J Appl Polym Sci. doi:10.1002/app.39628
Huang HD, Ren PG, Xu JZ, Xu L, Zhong GJ, Hsiao BS, Li ZM (2014) Improved barrier properties of poly(lactic acid) with randomly dispersed graphene oxide nanosheets. J Membr Sci 464:110–118
Nandi D, Talukdar A, Ghosh UC, De A, Bhunia S, Ghosh AK (2012) Reduced graphene oxide/ethylene vinyl acetate co-polymer composite with improved thermal stability and flame retardancy. J Polym Mater 29(4):401–411
Cui Y, Kundalwal SI, Kumar S (2016) Gas barrier performance of graphene/polymer nanocomposites. Carbon 98:313–333
Xu WB, Liag G, Wang W, Tang S, He P, Pan WP (2003) PP-PP-g-MAH-Org-MMT nanocomposites. I Intercalation behavior and microstructure. J Appl Polym Sci 88(14):3225–3231
Wang Y, Chen FB, Li YC, Wu KC (2004) Melt processing of polypropylene/clay nanocomposites modified with maleated polypropylene compatibilizers. Compos Part B Eng 35(2):111–124
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
ESM 1
(DOC 249 kb)
Rights and permissions
About this article

Cite this article
Doufnoune, R., Haddaoui, N. Effects of surface functionalized partially reduced graphene oxide and different compatibilizers on the properties and structure of PP/EPR nanocomposites. J Polym Res 24, 138 (2017). https://doi.org/10.1007/s10965-017-1302-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-017-1302-8