
Abstract
New methacrylate monomers 2-{[4-(dimethylamino) phenyl] amino}-2-oxoethyl-2-methylacrylate (DMPAMA), and 1-[4-(prop-2-yn-1-yloxy) phenyl] ethanone-O-methacryloyloxime (POEMO) were synthesized first time. The free radical copolymerization of DMPAMA with POEMO has been carried out in 1, 4-dioxane solution at 65 °C ± 1 using 2,2′-azobisisobutyronitrile (AIBN) as an initiator with different monomer-to-monomer ratios in the feed. 1H-NMR was used to determine the molar fractions of DMPAMA and POEMO in the copolymers. The monomer –reactivity ratios were calculated according to the general copolymerization equation using Kelen-Tüdõs and Finemann-Ross linearization methods. The polydispersity indices of the polymers determined with gel permeation chromatography. The thermal behaviors of copolymers with various compositions were investigated by differential scanning calorimetry and thermogravimetric analysis. The copolymer has been converted into a novel salts by reaction with the iodemethane (CH3I). The electrical conductivity dependence of temperature of the polymers were measured and the polymers exhibit the semiconducting behavior, confirming that the electrical conductivity increases with increasing temperature. All the products showed moderate activity against different strains of bacteria and fungi. In addition, photo-stability tests of the polymers under near-UV irradiation were performed.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Several studies have been done in our laboratories on the synthesis of new methacrylates and their radical commercially available monomers [1–4]. The use of homopolymers and copolymers especially designed with functional active groups as lateral substituents of the main chain is a topic increasing activity and interest. Copolymerization is the most successful method adopted for the preparation of materials with tailor made properties [5, 6].
Methacrylate polymers are among the most important commercial polymers, with a very widerange of applications in products as diverse as glazing, lighting housings, bath tubs and structural adhesives. The success of these polymers in many of the applications is dependent on the versatility of the acrylic monomers in copolymerizing to produce a wide diversity of structures, which can be tailored to produce the desired properties. Of particular interest is the synthesis of copolymers with specific functionality, which can act as a locus for further reactions, or furnish the copolymer with specific chemical and/or physical characteristics.
Recent investigations report the use of oxime esters as irreversible acyl transfer agents where the leaving group, the oxime does not participate in the back reaction [7]. This methodology has been elegantly utilized for the preparation of chiral polymers [8], regioselective acylation of nucleosides and to obtain various nucleoside derivatives of medicinal significance [9]. In a previous report [10], methacrylate containing oxime ester moieties used as irreversible acyl transfer agents.
Alkynes are important skeleton in some natural products, pharmaceuticals, biologically active molecules, and nonlinear optical materials. [11, 12]. For example, compounds AGN-193,109 and AGN-1,943,101 containing the biaryl alkynes moieties are RAR antagonists for the treatment of mucocutaneous toxicity currently in Phase III clinical trials [13]. Terminal alkynes used in recent years extensively in the click chemistry.
Quaternary ammonium salts (QAS) are extensively used in various applications. They are present in fabric softeners and corrosion inhibitors [14, 15], they act as fungicides, pesticides and insecticides [16], they reveal antibacterial and antifungal activities employed in antimicrobial drugs [17–19], they are ingredients of shampoos and hair conditioners [20–22] and they are used in organoclay preparations, which have diverse applications, including environmental remediation and synthesis of nano-materials [23–25].
Knowledge of the copolymer composition is an important step in the evaluation of its utility. The copolymer composition and its distribution depend on monomer reactivity ratios. The most common mathematical model of copolymerization is based on finding the relationship between the composition of copolymers and the composition of the monomer feed in which the monomer-reactivity ratios are the parameters to be determined [26, 27]. The calculation of the monomer -reactivity ratios requires the mathematical treatment of experimental data on the composition of copolymers and monomer in feed mixtures. The most fundamental quantity characterizing a copolymer is its composition on a molar basis, which eventually is used for the determination of the relevant monomer reactivity ratios. Spectroscopic methods, preferably 1H NMR and 13C NMR spectroscopy, [28, 29] and elemental analysis are probably the most widely used methods for the analysis of copolymers and the determination of reactivity ratios r1 and r2.
Thermogravimetric analysis (TGA) has been widely used to investigate the decomposition characteristics of many materials. Some methods have already been established to evaluate the kinetic parameters from thermogravimetric data [30, 31].
POEMO and DMPAMA are new functional methacrylic monomer with side chain alkynes and tertiary amine group respectively. In this work, the results of the radical copolymerization of DMPAMA with POEMO, the determination of the monomer reactivity ratios with 1H-NMR analysis.
Experimental section
Materials
4-Hydroxyacetophenone, methacryloyl chloride, propargyl bromide, chloroacetyl chloride, sodium methacrylate, N,N-dimethyl-p-phenylenediamine and hydroxyl amin hydrochloride (Sigma) were used as received. Ethanol, methanol, chloroform, n-hexane and benzene were freshly distilled over Molecular Sieves prior to use. 1, 4-dioxane, acetonitrile and potassium carbonate (Sigma) were used as received. Azobisizobutyronitrile (AIBN) was recrystallized from a chloroform-methanol mixture. All the other chemicals were analytical-grade, and they were used without any further purification.
Measurements
Infrared spectra were obtained with a Perkin Elmer BXII infrared (FTIR) spectrometer with KBr pellets in the 4,000–400 cm-1 range, and 10 scans were taken at a 4 cm−1 resolution. 1H NMR and 13C NMR spectra in DMSO solutions were recorded on a Bruker GmbH 500 MHz FT-NMR spectrometer with tetramethylsilane as an internal reference. The Mw and Mn value of the polymer were determined with a Waters 410 gel permeation chromatograph equipped with a refractive index detector and calibrated with polystyrene standards. Thermal data were obtained with a Shimadzu DTA-DSC-60H instrument at a heating rate of 10 °C min−1 in an N2 atmosphere.
Electrical conductivity was measured as a function of temperature by alternating polarity methods to eliminate electrical polarization, triboelectric and piezoelectric effects using a KEITHLEY 6517A electrometer.
Synthesis of the 1-[4-(prop-2-yn-1-yloxy) phenyl] ethanone
Synthesis of 1-[4-(prop-2-yn-1-yloxy) phenyl] ethanone was as follows: p-hydroxy acetophenon (1 mol ) and K2CO3 (1 mol) were dissolved in 20 ml of anhydrous acetonitrile at 25 °C, and then propargyl bromide (1 mol) was added dropwise to the solution. The reaction mixture was stirred at 70 °C, for 4 h. The organic layer was washed several times with diethyl ether and dried over MgSO4. Yield: 85 %.
IR (neat),cm−1:1,700 (C = O for carbonyl), 1,610 (aromatic C = C), 1,250 (-C-O-C-), 3,290 and 2,121 (HC ≡ and -C ≡ C-). The reaction path are shown in Scheme 1.

Synthesis of the 1-[4-(prop-2-yn-1-yloxy) phenyl] ethanone
Synthesis of the 1-[4-(prop-2-yn-1-yloxy) phenyl] ethanone oxime
Synthesis 1-[4-(prop-2-yn-1-yloxy) phenyl] ethanone oxime was synthesized according to standard method [32]. The synthesis route is shown in the Scheme 2. The yield was: 83 %.

Synthesis of the 1-[4-(prop-2-yn-1-yloxy) phenyl] ethanone oxime
IR (neat), cm−1: 3,300-3,400 (OH stretching for oxime ), 1,610 (aromatic C = C), 1,250 (-C-O-C-), 3,100 (aromatic C-H), 3,290 and 2,121 (HC ≡ and -C ≡ C-). The reaction path are shown in Scheme 1.
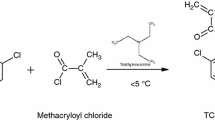
Synthesis of the POEMO monomer
1-[4-(prop-2-yn-1-yloxy) phenyl] ethanone-O-methacryloyloxime (POEMO) was prepared as reported.[33] The synthesis of monomer is shown in Scheme 3. The yield was about 85 %.

Synthesis of POEMO monomer
IR (KBr), cm−l :1,775 ( methacrylic carbonyl), 1,635 (CH2 = C-), 1,600 (C = C), 1,500 (C = N) 1,254 (Ar-O-CH2-), 3,290 and 2,121 (HC ≡ and -C ≡ C-).
1H-NMR (δ, ppm from TMS in CDCl3): 7.0–8.3 (aromatic protons, 4H); 5.6 (CH2=, 1H); 6.2 (CH2=, 1H); 5.3 (-OCH2-, 2H); 2.3 (-N = CCH3, 3H); 3.2 (HC≡); 1.9 (-CH3 ,3H).
13C-NMR (δ, ppm from TMS in CDCl3): 169.0 (C = O of esters ); 130.0 (=C ); 129.1 (CH2=); 114.1–159.8 ( aromatic carbons); 65.0 (-OCH2-); 16.5 (N = CCH3); 78 (HC≡), 79 (C≡); 18 (CH3).
Synthesis of the DMPAMA monomer
2-{[4-(dimethylamino) phenyl] amino}-2-oxoethyl-2-methylacrylate (DMPAMA) was prepared as reported [34]. The monomer was examined by FT-IR, 1H and 13C NMR spectra. The synthesis of monomer is shown in Scheme 4. Yield: 82 %.

Synthesis of DMPAMA monomer
IR (KBr pellet), cm−1; 3,100 (Ar-H stretching); 1,725 and 1,680 (ester and amide carbonyl); 1,630 (CH3-C = CH2); 1,254 (C-O-C); 2,960 (Aliphatic C-H).
1H-NMR (δ, ppm from TMS in CDCl3): 6.8–7.5 (aromatic protons, 4H); 5.6 (CH2=, 1H); 6.2 (CH2=, 1H); 4.7 ( OCH2); 2.8 [-N (CH3) 2] ; 1.9 (-CH3).
13C-NMR (δ, ppm from TMS in CDCl3): 169 (amide carbonyl); 167 (ester carbonyl); 135 (C= ); 126 (CH2=); 66 (OCH2); 42 [-N (CH3) 2]; 117–144 (Ar-C).
Synthesis of the homopolymers
Polymerization of monomers were carried out in glass ampoules under N2 atmosphere in 1,4-dioxane solution with AIBN (1 % based on the total weight of monomers) as an initiator. The reacting components were degassed by three freeze-thawing cycles and then immersed in oil bath at 65 °C for a given reaction time. The polymers were separated by precipitation in ethanol and reprecipitated from CH2Cl2 solution. The polymers were finally dried under vacuum to constant weight at room temperature and kept in a desiccators under vacuum until use.
Copolymerization
Copolymerization of DMPAMA with POEMO, with seven different feed compositions, were carried out in 1, 4-dioxane at 65 °C with AIBN (1 %, based on the total weight of the monomers) as an initiator. Appropriate amounts of DMPAMA with POEMO and 1, 4-dioxane was mixed in a polymerization tube, purged with N2 for 20 min, and kept at 65 °C in a thermostat. The reaction time (≈5 h) was selected to give conversions less than 10 wt.% to satisfy the differential copolymerization equation. The conversion of the monomer to the polymer was determined by a gravimetric method. After the desired time, the copolymerization was stopped. These copolymers were poured into excess ethanol to precipitate, were filtered, were purified by repeated reprecipitation from a solution of each polymer in 1, 4-dioxane by ethanol, and were dried in a vacuum oven at 50 °C to a constant weight. The amounts of monomeric units in the copolymers were determined by 1HNMR. The constituent monomeric units of the copolymer are shown in Scheme 5.

Synthesis of poly (DMPAMA-co-POEMO)
Determination of the monomer reactivity ratios
The monomer reactivity ratios for the copolymerization of DMPAMA with POEMO were determined from the monomer feed ratios and the copolymer composition. The Fineman–Ross (FR) [35] and Kelen–Tudos (KT) [36] methods were used to determine the monomer reactivity ratios. Results from the analysis for FR and KT are presented in Table 3. According to the FR method, the monomer reactivity ratios can be obtained as follows:
where r1 and r2 correspond to the DMPAMA and POEMO monomers, respectively. The parameters G and H are defined as follows:
with
where M 1 and M 2 are the monomer molar compositions in the feed and m 1 and m 2 are the copolymer molar compositions.
Alternatively, the reactivity ratios can be obtained with the KT method, which is based on the following equation:
where η and ξ are functions of the parameters G and H
and α is a constant equal to (H max.H min) 1/2, H max and H min being the maximum and minimum H values, respectively, from the series of measurements. From a linear plot of η as a function of ξ, the values of η for ξ = 0 and ξ =1 can be used to calculate the reactivity ratios according to the following equations:
Antimicrobial activity of the polymers
The biological activities of the monomers and their homopolymers and copolymers were tested against different microorganisms with DMSO as the solvent. The sample concentrations were 100 μg. In this study, Staphylococcus aureus ATCC 29,213, Escherichia coli ATCC 25,922, Pseudomonas aeruginasa ATCC 27,853, proteus vulgaris, Salmonella enteridis, and Klebsiella pneumoniae were used as bacteria. Candida albicans CCM 31 was a fungus. YEPD medium cell culture was prepared as described by Connerton [37]. Ten milliliters of YEPD medium were inoculated with each cell from plate cultures. Yeast extract 1 % (w/v), bactopeptone 2 % (w/v), and glucose 2 % (w/v), was obtained from Difco. Microorganisms were incubated at 35 °C for 24 h. About 1.5 ml of these overnight stationary phase cultures were inoculated onto 250 ml of YEPD and incubated at 35 °C until OD600 reached 0.5.
The antibiotic sensitivity of the polymers was tested with the antibiotic disk assay as described [38]. Nutrient Agar (NA) was purchased from Merck. About 1.5 ml of each prepared different cell culture were transferred into 20 ml of NA and mixed gently. The mixture was inoculated into the plate. The plates were rotated firmly and allowed to dry at room temperature for 10 min. Prepared antibiotic discs (100 μg) were placed on the surface of the agar medium [39]. The plates were kept at 5 °C for 30 min and then incubated at 35 °C for 2 days. If a toxic compound leached out from the disc, it means that the microbial growth is inhibited around the sample. The width of this area expressed the antibacterial or antifungal activity by diffusion. The zones of inhibition of microorganism growth of the standard samples monomers, homopolymers and copolymers, were measured with a millimeter ruler at the end of the incubation period.
Quaternization of the copolymers
The prepared poly (DMPAMA-co-POEMO) (0.53:0.47) ionomer was immersed in a mixture of iodomethane and acetonitrile (1/2, v/v) at 25 °C. Stirring was continued for 12 h to ensure the full reaction. After reacting for 12 h, the mixture were precipitated in n-hexane and washed by ethylacetate. The products were dissolved in dichloro methane and precipitated in the ethylacetate and then dried at 50 °C. The quaternization ratio (QR) of copolymers were obtained according to an equation as following [40]:
where W0 is the mass of the poly (DMPAMA-co-POEMO) before quaternization, W1 is the mass of the poly (DMPAMA-co-POEMO) after quaternization 519 is the molecular weight of poly (DMPAMA-co-POEMO) unit, and 142 is the molecular weight of methyl bromide. QR values of all the copolymers was about 1 after 12 h. The chemical structure of the ionomer can be represented according to Scheme 6:

Synthesis of the ionomers
Results and discussion
Characterization of the homopolymers
In the FTIR spectrum of the poly (POEMO) showed some characteristic absorption peaks at 1,725 cm−1 (oxime ester carbonyl stretchings) and at 3,204 and 2,117 cm−1 ( HC ≡ and -C ≡ C-). During the polymerization of the monomers, the IR band at 1,633 cm−1 (C = C) disappeared and ester carbonyl stretchings for polymers shifted to about 1,742 cm−1. The main evidence of the polymer is certainly the disappearance of some characteristic signals of the double bond in the spectra and this fact was effectively observed in our case. Thus, two bands vanished in the IR spectrum: the absorption band at 923 cm−1 assigned to the C-H bending of geminal = CH2 and the stretching vibration band of C = C at 1,600 cm−1. The 1H-NMR spectrum of poly (POEMO) is shown in Fig. 1. Disappearance of two 1H NMR singlets of vinyl protons at 5.6 and 6.2 ppm and appearance of broad resonance at 1.5 and 2.2 ppm due to aliphatic protons give clear indication of the formation of polymers. In the proton decoupled 13C NMR spectrum of poly (POEMO), chemical shift assignments were made from the off-resonance decoupled spectra of the polymer. Resonance signals at 176 ppm correspond to ester group present in polymer. The α-methyl group of polymer shows resonance signals at 14. The aromatic carbons of the both polymers were observed between 120 and 160 ppm. The poly (DMPAMA) is characterized with same methods. The 1H-NMR and 13C-NMR spectrum of the poly (POEMO) are in good agreement with the structure.

The 1H-NMR spectrum of the poly (POEMO)
Characterization of the copolymers
Solubility
The solubility of the homopolymers and copolymers was tested via the mixing of 10 mg of each
polymer with 3 ml of various solvents in test tubes. After the closed tubes were set aside for 1 day, the solubility was observed. The homopolymers and copolymers were soluble in 1, 4-dioxane, dichloromethane, dimethylacetamide, dimethylformamide, dimethyl sulfoxide, tetrahydrofuran, but were insoluble in n-hexane, n-heptane, ethanol, and methanol solvents.
FTIR spectrum
In the FTIR spectrum of the copolymer, the peaks at about 3,204 and 2,117 cm−1 are attributed to HC ≡ and -C ≡ C- groups respectively. The peak at 3,095 cm−1 corresponds to the C–H stretching of the aromatic system. The symmetrical and asymmetrical stretching due to the methyl and methylene groups are observed at 2,980, 2,940 and 2,866 cm−1. The peaks at about 1,739 cm−1 are attributed to the ester carbonyl stretchings of DMPAMA and POEMO units. The amide carbonyl peak of DMPAMA units are seen at 1,678 cm−1. The asymmetrical and symmetrical bending vibrations of methyl groups are seen at 1,462 and 1,383 cm−1. The C–O stretching is observed at 1,161 and 1,210 cm−1. The C–H and C−C out of plane bending vibrations of the aromatic nuclei are observed at 792 and 561 cm-1, respectively.
1H NMR spectrum
The 1H NMR spectrum of the copolymer poly (DMPAMA-co-POEMO) (0.52:0.48) is shown in Fig. 2a. The chemical shift assignments for the copolymers were based on the chemical shifts observed for the respective homopolymers. The aromatic protons show signals between 6.72 and 7.35 ppm. The spectrum shows a signal at 4.6 and 4.8 ppm, which are due to –OCH2– group at POEMO and DMPAMA units respectively. The backbone methylene groups show signals at 2.04–2.20 ppm. The signals obtained at 1.10 and 1.22 ppm are due to the α-methyl protons of POEMO unit. The methyl protons of the DMPAMA and POEMO units show signals 2.62 ppm. The acetylenic proton on the POEMO units is shown at 3.22 ppm. The NH proton on the DMPAMA units is shown at 9.84 ppm.

a 1H NMR and b 13C-NMR Spectra of poly (DMPAMA-co-POEMO) (0.53:0.47)
13C NMR spectrum
The proton decoupled 13C NMR spectrum of the copolymer poly (DMPAMA-co- POEMO) (0.52:0.48) is shown in Fig. 2b. The two ester carbonyl of POEMO appeared at 166 and 168 ppm. The amide carbonyl signal of DMPAMA units are observed at 172 ppm. The aromatic carbons in copolymer appeared at 115–150 ppm. The methylenoxy groups in POEMO and DMPAMA units show signal between 66 and 69 ppm. The signals due to the backbone methylene carbon atoms are observed at 43.2 ppm. The α-methyl carbon atoms of DMPAMA and POEMO units give a series of resonance signal at 18.2 ppm. The methyl carbons on the nitrogne atom of DMPAMA units are shown at 42 ppm. The acetylenic carbons of POEMO units are shown at 79 ppm. The peak at 152 ppm are attributed azomethine carbons (C = N) of POEMO units.
Copolymer compositions and monomer reactivity ratios
The copolymerization of DMPAMA with POEMO in a 1, 4- dioxane solution was studied for POEMO molar fractions of approximately 0.80–0.25 in the feed. The number of monomeric units in the copolymers was determined by 1H-NMR spectroscopy analysis. The assignment of the resonance peaks in the 1H NMR spectrum leads to accurate evaluation of each monomeric content incorporated into the copolymer chains. Thus, the mole fraction of DMPAMA in the copolymer was calculated by measuring the integrated peak height of total methyl protons to that of total aromatic protons in the copolymer. The following expression is used to determine the composition of the copolymers. Let m1 be the mole fraction of DMPAMA and 1 -m1 be that of POEMO. POEMO contains four aromatic protons and two methyl group due to nitrogen (six proton), and DMPAMA contains four aromatic protons and three methyl protons. The following expression is used to determine the composition of copolymers.
On simplification
The molar fractions of DMPAMA in the copolymers were determined by with Eq. 2. The results are presented in Table 1.
Fineman-Ross (FR), and Kelen-Tüdös (KT) methods were used to determine the monomer reactivity ratios. The graphical plots concerning the methods previously reported are given in Fig. 3a, b. The values are presented as follows:

a FR plots and b KT plots for determining the monomer reactivity ratios in the copolymerization of DMPAMA (M 1) and POEMO (M 2)
Methods | r 1 | r 2 | r 1 r 2 | 1/r 1 | 1/r 2 |
F-R | 0.56 | 0.36 | 0.20 | 1.79 | 2.78 |
K-T | 0.78 | 0.45 | 0.35 | 1.28 | 2.22 |
In all cases and for all graphical methods, the plots are linear, and this indicates that these copolymerizations follow conventional copolymerization kinetics and that the reactivity of a polymer radical is determined only by the terminal monomer unit. The higher r 1 values of DMPAMA confirm its higher reactivity compared with that of POEMO. The higher fraction of DMPAMA in the copolymers indicates that the probability of DMPAMA entry into the chain is greater than that for POEMO. Thus, the copolymers formed are richer in DMPAMA.
However, in these copolymerizations the copolymer sequences will be an alternating tendency, since the low values of rPOEMO and rDMPAMA.
Molecular weights of the polymers
The molecular weights of the polymers were determined by GPC with polystyrene and tetrahydrofuran as the standard solvent, respectively. The Mw and Mn values and polydispersity indices (Mw/Mn) of the polymer samples are presented in Table 2. The polydispersity index of the polymers ranges from 1.68 to 1.85. The theoretical values of Mw/Mn for the polymers produced via radical recombination and disproportionation are 1.5 and 2.0, respectively. The values of Mw/Mn in copolymerization are also known to depend on chain termination in the same way as in homopolymerization.[41].
Glass transition temperatures
The glass-transition temperatures (Tg’s) were determined with a Shimadzu DSC-60 instrument. From DSC measurements, Tg was taken as the midpoint of the transition region. All the copolymers show a single Tg, and this shows the absence of a mixture of homopolymers or the formation of a block copolymer. Tg of poly (POEMO) is 79 °C, and that of poly poly (DMPAMA) is 97 °C. The Tg values of the copolymers increase with an increase in the DMPAMA content in the copolymers. The results clearly indicate that the Tg values of the copolymers depend on the compositions of the comonomers and increase with increasing DMPAMA content in the polymer chain. Plots of the Tg’s of the copolymers versus copolymer composition are shown in Fig. 4. Tg values for the DMPAMA–POEMO copolymers depend linearly on their compositions. This trend in the data indicates the ideal mixing of the two components in this copolymer system, and the specific volumes for the copolymers are similar to that for ideal mixing. The Tg values of the copolymers are indicated in the Table 3.

Variation of Tg (°C) with composition of poly (DMPAMA-co-POEMO) system
The thermal properties of the copolymers are influenced by their chemical structure and composition and the monomer sequence distributions. Several relationships have been employed to describe the effect of these parameters on the glass transition temperature of the copolymers. The simplest equation describing the effect of composition on Tg is the Gibbs–Di Marzio equation [42]:
where Φ DMPAMA, Φ POEMO are the mole fractions of DMPAMA and POEMO monomers respectively in the copolymer and Tg DMPAMA , Tg POEMO the glass transition temperatures of the two homopolymers respectively.
A similar relationship was introduced by Fox [43]:
where W DMPAMA and W POEMO are the weight fractions of DMPAMA and POEMO monomers in the copolymer. The experimental results concerning the Tg of the DMPAMA and POEMO copolymers along with the predictions of the Gibbs–Di Marzio and Fox equations are given in Table 3. It is obvious that large positive deviations are obtained by these two methods due to the fact that they are based only on thermodynamic and free volume theories of the glass transition and they do not take into consideration the monomer sequence distribution and the effect of their compatibility on steric and energetic interactions. Therefore several models have been proposed that take into account these considerations.
Thermogravimetric analysis
TGA curves for poly (DMPAMA), poly (POEMO) and two copolymers are shown in Fig. 5. The initial decomposition temperature (IDT) of poly (DMPAMA) and poly (POEMO) are 285, and 220 °C. The thermal stability of poly (DMPAMA) is greater than the poly (POEMO). The reason for this may be hydrogen bonds between the molecules of poly (DMPAMA). The degradation of poly (DMPAMA) occurred in two stages. The first stage was observed at 280–335 °C. The second stage decomposition commenced at 376–445 °C. The degradation of poly (POEMO) occurred in two stages. One stage at 225–325 °C and two stage at 360–425 °C are observed. The residue at 450 °C of poly (DMPAMA) and poly (POEMO) are 24 % and 34 %, respectively.

TGA curves for homopolymers and some copolymers
The DTG curves of copolymers showed that the thermal decomposition took place mainly in two stages and it is understood from TG/DTG curves, the first and second decomposition temperatures for copolymers are in range of 235–410 °C and 385–475 °C, respectively. Some degradation characteristics of the copolymers are given in Table 4 by comparison with those of the homopolymers. The thermal stabilities of copolymers are between those of the corresponding homopolymers. The thermal degradation of poly (n-alkyl methacrylate) s typically produces the monomer as a result of depolymerization [44, 45]. The formation of cyclic anhydride- type structures by intramolecular cyclization is another main process in the degradation of these polymers. The latter produces some low-molecular-weight products, depending on the chemical structures of the side chain of poly (methacrylic ester) s.
Antimicrobial screening
The antimicrobial activity of polymers in general increases with increasing hydrophobic content in the side chains. This is likely because of the increased hydrophobicity of polymers enhancing insertion of polymers into the hydrophobic region of cell membranes. Our recent investigation on methacrylate polymers indicated that the hydrophobic side chains of methacrylate random copolymers are inserted into lipid bilayers [46].
All the compounds exhibited good activity comparable to that of the standard drugs. The data reported in Table 5 are the average data of three experiments. The results show that the investigated polymers have good biological activity comparable to that of standard drugs such as penicilin, g. and teicoplanin. Poly (DMPAMA) have more activity than that of poly (POEMO). The activity of the copolymers are between of homopolymers. The activity of the copolymers against microorganism increased with increasing DMPAMA content in the copolymers. The results suggest that the monomers, polymers and the some copolymers have good biological activity on the pseudomonas aeruginasa and esherichia coli microorganisms. It can be seen from Table 5 that the antibacterial effect of copolymers are less to that of the standards. Further the antibacterial and antifungal actions of copolymers may be significantly enhanced teriary amine groups of DMPAMA and alkynes groups of POEMO. It has been suggested that poly (DMPAMA), poly (POEMO) and its copolymers with N, and O donor systems and the carbonyl and oxime ester groups might have inhibited enzyme production because enzymes that require a free hydroxy group for their activity appear to be especially susceptible to deactivation by the donor atoms of the polymers. The mechanism of action of antimicrobial polymers is different from that of monomeric antimicrobial agents. While monomeric gents diffuse into the cells or accumulate in the cell walls, polymers affect the central membrane functions of the microorganism from the outside by electrostatic interaction with the cell wall. Antimicrobial effect is unspecific and includes Gram-positive and gram-negative bacteria, fungi and yeasts [47]. This contact disrupts transportation through its membrane or cause damage to the cell wall.
Characterization of Ionomer by 1H-NMR Spectra
The 1H-NMR spectrum of poly (DMPAMA-co-POEMO) (0.53:0.47) ionomer is shown in Fig. 6. The chemical shift assignments for the ionomers were based on the chemical shifts observed for the respective copolymers. The structure of ionomer is confirmed by 1H-NMR spectral data.

1H-NMR spectra of poly (DMPAMA-co-POEMO) (0.53:0.47).ionomer
Semi-conducting Properties of the Poly (DMPAMA-co-POEMO) Ionomers
Figure 7 shows the electrical conductivity plots of the ionomer. The electrical conductivity of the poly (DMPAMA-co-POEMO) (0.53:0.47) polymer doped by CH3I increases with an increase of temperature. The electrical conductivity of copolymers were fitted by Arrhenius model:

Electrical conductivity plots of the poly (DMPAMA-co- POEMO) (0.53:0.47] ionomer
Where σ0 is a constant, ΔE is the activation energy for conductivity, T is the temperature. As seen in Fig. 7, the poly (DMPAMA-co-POEMO) (0.53:0.47) polymer doped by CH3I for 12 h. shows the highest conductivity. The ΔE and room temperature conductivity values of this polymer were obtained to be 0.139 eV and 3.6 × 10−7 S/cm, respectively. These electronic parameters suggest that the poly (DMPAMA-co-POEMO) (0.53:0.47) polymer doped by CH3I for 12 h is an organic semiconductor and the conductivity mechanism is controlled by the thermally activated conduction mechanism. It is evaluated that the electrical conductivity properties of the insulating poly (DMPAMA-co-POEMO) (0.53:0.47) changed from insulating state to semiconducting state using methyl iodide dopant.
Photophysical properties
A solution of polymers in CH2Cl2 was cast onto a quartz glass plate with a spin coater and then was annealed for 30 min at 40 °C. The polymer films were irradiated with a 500-W, high-pressure mercury lamp at 254 nm. The films of polymers were exposed to UV light and, as expected, all of them presented alteration in the FTIR spectra as a function of time. The discussion of the results will focus on the absorption evolution in the C = O stretching range (assigned to carbonyls), close to 1,730 cm−1. After irradiation for 12, 24, 36, and 72 h, the FTIR spectra of the polymers showed changes. A new band appearing as a shoulder at 1,705 cm−1 was observed in the spectra of the polymers, and the absorbance of the shoulder at 1,705 cm−1 increased relatively as that of the band at about 1,730 cm−1 decreased from 12 to 72 h. In addition, although all of the polymers were soluble in CH2Cl2, CHCl3, dimethylformamide, DMSO, 1,4-dioxane, and so forth before irradiation, not one of the polymers was soluble in any solvent. Although a practical evaluation from spectral data is rather dubious because the sensitivity of photocrosslinkable polymers is a function of the Tg, molecular weight, polymer solubility, and so forth, the discrimination of photoresponsibilities of the chromophores themselves is possible to a certain extent. To achieve a highly integrated chromophore system, an arrangement of the chromophores in a first-order structure of a polymer is not enough; one should resort to a higher order structure formed by the polymer [48]. In this context, a number of studies have focused on amphiphilic polyelectrolytes carrying aromatic chromophores as pendant groups [49]. The amphiphilic polyelectrolyte loadings are high enough that the hydrophobic chromophores are forced to be densely packed in an aqueous solution [50]. All the experimental data indicate that photodegradation in the carbonyl region occurred and crosslinking followed. Some polymers with carbonyl and oxime esters side chains displayed photodegradation [51].
Conclusion
The synthesis of new methacrylate monomers (DMPAMA) and (POEMO) having pendant tertiary amine and alkynes moieties have been reported for the first time. The structure of monomers and its homopolymers were characterized by spectroscopic methods. Copolymers of DMPAMA with POEMO were prepared by free-radical polymerization in 1,4-dioxane at 65 °C. The reactivity ratios of the copolymers were estimated with linear graphical methods. The r1 values were higher than the corresponding r2 values in all cases, and this means that a kinetic preference exists for the incorporation of DMPAMA in the copolymer structure. The values strongly suggest that the growing radicals of both monomeric ends preferentially add to the DMPAMA monomer, thus leading to the formation of a copolymer with a higher amount of DMPAMA. GPC data imply that the polydispersity index of the copolymers is nearly equal to 2, and this implies a strong tendency for chain termination by disproportionation. Tg of the copolymers increased with increasing POEMO content in the copolymers. The some copolymers have good biological activity on the pseudomonas aeruginasa and esherichia coli microorganisms. The electrical conductivity properties of the insulating copolymers changed from insulating state to semiconducting state using methyl iodide dopant. Finally, it was observed that most the synthesized polymers are not stable under near-UV irradiation.
References
Erol I, Sarkaya S (2012) J Polym Res 19:9957
Erol I, Yavuz F (2005) Polym Int 54(3):506
Soykan C, Erol I (2003) Euro Polym J 9(12):2261
Erol I, Soykan C, Ahmedzade M (2002) J Polym Scı Part A-Polym Chem 40(11):1756
Demirelli K, Coskun M, Erol I (2000) Euro Polym J 36(1):83
Vijayanand PS, Kato S, Satokawa S, Kojima T (2009) J Polym Res 16(3):301
Johnson A, Wang L, Standeve A, Escobar M, Chandraratna R (1999) Bioorg Med Chem 7:1321
Binks BP, Fletcher PD, Salama IE, Horsup DI, Moore JA (2011) Langmuir 27:469
Singh S, Bhadani A, Kataria H, Kaur G, Kamboj R (2009) Ind End Chem Res 48:1673
Zielinski R (ed) (2005) Zielinski. R Quaternary Ammonium Salts, ITD Press, Poznan
Rabea EI, Badawy MEN, Stevens CV, Smagghe G, Steurbaut W (2003) Biomacromolecules 4:1457
Belalia R, Grelier S, Benaissa M, Coma V (2008) J Agric Food Chem 56:1582
McDonnell JG, Russell AD (1999) Clin Microbiol Rev 12:147
Boethling RS (1984) Water Res 18:1061
Levinson MI (1999) J Surfactants Deterg 2:223
22.Cross J, Singer E.J (1994) Marcel Dekker Inc New York .
Sarkar B, Xi Y, Megharaj M, Krishnamurti GS, Naidu R (2010) J Colloid Interface Sci 350:295
Sarkar B, Xi Y, Megharaj M, Krishnamurti GS, Naidu R (2010) J Hazard Mater 183:87
Sarkar B, Xi Y, Megharaj M, Krishnamurti GS, Naidu R (2011) Appl Clay Sci 51:370
Arshady R, Kenner GW, Ledwith A (1981) Macromol Chem Phy 182(1):41
27.Ham G (1964) High Polymers Interscience Vol 18 New York.
Erol I, Ahmedzade M (2005) J Polym Res 12(4):247
Bednarski R, Braun D (1990) Makromol Chem 191(4):773
Chang TC, Shen WY, Chiu YS, Chen HB, Ho SY (1996) J Polym Sci Part A: Polym Chem 34(16):3337
Chang TC, Shen WY, Chiu YS, Chen HB, Ho SY (1997) Polym Degrad Stab 57(1):7
32.Vogel A (1989) Vogels Textbook of Pratical Organic Chemistry Longman p.813
Erol I, Kolu S (2011) J App Polym Sci 120(1):279
Erol I, Dedelioglu A (2008) Journal of Polymer Scıence Part. A-Polymer Chemstry 46(2):530
Fineman M, Ross SD (1950) J Polym Sci 5(2):259
Kelen T, Tudos F (1975) J Mac Sci Pure Appl Chem 9:1
37.Connerton I.F, In Analysis of Membrane Proteins, Ed by Gould G.W., Portland, London, 177(1994).
Chan ECZ, Pelczar MJ, Krieg NR (1993) Mc-Graw-Hill. New York p 225
Desai JA, Dayal U, Parsania PH (1996) J Macromol Sci Pure Appl Chem 33(8):1113
Yang YF, Hu HQ, Li Y, Wan LS, Xu ZK (2011) J Membr Sci 376:132
Melville HW, Noble B, Watson WF (1649) J Polym Sci 4((5):629
Gibbs JH, Di Marzio EA (1963) J Polym Sci 1:1417
Fox TG, Flory PJ (1950) J Appl Phys 21:581
Erol I, Soykan C, Ilter Z, Ahmedzade M (2003) Polym Degrad Sta 81:287
Coskun M, Erol I, Coskun MF, Demirelli K (2002) Polym Degrad Stab 78:49
Erol I (2008) J Fluorine Chem 129(7):613
Munoz-Bonilla A, Fernandez-Garcia M (2012) Progress Polym Scı 37(2):281
Guillet JE, Rendall WA (1986) Macromolecules 19:224
Morishima Y, Itoh Y, Nozakura S (1981) Makromol Chem 82:3135
Morishima Y, Kobayash T, Nozakura S (1989) Polym J 21:267
Weir NA, Arct J (1990) Whiting K. Eur Poly J 26:341
Ghogare A, Kumar S (1989) J Chem Soc Chem Com 5:1533
Ghogare A, Kumar S (1989) J Chem Soc Chem Com 5:134
Moris F, Gotor V (1993) J Org Chem 58:653
Athawale V, Manjrekar NJ (2000) Mol Catal B Enzymatic 10:551
Bohlmann F, . Burkhart F.T, Zero C (1973) London New York.NY.
Hunstman V.D, Patai S (1978) Wiley Interscience London 553
Acknowledgments
Author wish to thank the financial support provided by the Afyon Kocatepe University Research Fund (Project No:11-FENBİL-02).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Erol, I., Özcakir, R. & Gürler, Z. Novel functional methacrylate copolymers with side chain tertiary amine and alkynes and their some properties. J Polym Res 22, 635 (2015). https://doi.org/10.1007/s10965-014-0635-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-014-0635-9