
Abstract
Poly(L-lactide) (PLLA) oligo-esters with α-hydroxyl-ω-alkyl (alkyl = −CH2−[CH2−CH2]m−CH3, where m = 1, 2, 4, 5, 6, 7, 8, 9, and 10) end groups were synthesized by ring-opening polymerization of L-lactide (L-LA) catalyzed by tin(II) 2-ethylhexanoate Sn(Oct)2 in the presence of aliphatic alcohols as initiators (HO−CH2−[CH2−CH2]m−CH3, where m = 1, 2, 4, 5, 6, 7, 8, 9, and 10). High yields (~ 62 to 71%) and M n(NMR) in the range of 2120–2450 Da (PLLA) were obtained. Effects of alkyl end groups on thermal properties of the oligo-esters were analyzed by DSC, TGA and SAXS. Glass transition temperature (T g) gradually decreases with increase in the percent of−CH2−[CH2−CH2]m−CH3 end group, as results alkyl end group provides most flexibility to PLLA. An important effect of alkyl end group on a double cold crystallization (T c1 and T c2) was observed, and is directly related with the segregation phase between alkyl end group and PLLA. TGA analysis revealed that PLLA oligo-esters are more thermally stable with docosyl (−C22H45) respect to the butyl (−C4H9) end group, probably is due to steric hindrance of the end group (docosyl respect to butyl) toward intermolecular and intramolecular transesterification. SAXS analysis showed that alkyl end group as docosyl restricted the growth of lamellae thickness (D) due to steric hindrance. Characterization of hydroxyl and alkyl end groups in the PLLA oligo-esters was determined by MALDI-TOF, GPC, FT-IR and 1 H and 13 C NMR.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Poly(L-lactide) (PLLA) 1 is an important aliphatic polyester with degradable properties and potentials application in biomedical area [1]. Typically, PLLA is obtained by ring-opening polymerization (ROP) of L-lactide (L-LA) in presence of a catalyst as tin(II) 2-ethylhexanoate or tin(II) octoate Sn(Oct)2 [1–3]. Sn(Oct)2 is the most important catalyst in ROP of lactones because was approved by Food and Drugs Administration (FDA) as food additive [4].

In ROP of lactones is common the use of initiators, for example: primary and secondary aliphatic alcohols (ROH) [2]. The initiator (ROH) can react with Sn(Oct)2 and produce a tin alkoxide (Oct-Sn-OR) by transfer reaction [5, 6], where tin alkoxide is active specie in the ROP of lactones as L-LA [6], as result a polyester with aliphatic (R) and hydroxyl (OH) end groups can be obtained. A similar synthetic method was reported by Seppälä and coworkers [3] in the synthesis of PLLA with different hydroxyl end groups.
PLLA with terminal groups α-hydroxyl-ω-alkyl has been used as: a) macroinitiator in the synthesis of diblock copolymers [7–9], b) compatibilizer within polymer blends polyethylene/PLLA [10, 11] and c) depressed factor in erosion surface of PLLA by enzymatic and hydrolytic degradation due to alkyl end group [12–14].
Recently, Abe and coworkers [12, 13] studied the effect of alkyl end groups as dodecyl (C12H27) or tetracosyl (C24H49) on enzymatic [12, 13] and nonenzymatic [13] degradation of the PLLA, they found that the alkyl end group affected the enzymatic degradation, end groups with carbon number bellow 11 and over 12 were homogeneously and heterogeneously degraded, respectively [13]. Contrary, the thermal properties of PLLA samples showed similar values and independent of the structure in the terminal alkyl group. In the two works previously reported [12, 13], only high molecular weights were used for the study of PLLA with different alkyl end groups (M n(GPC) = 106000 [12], and 73000 [13]). In a third work [14], the thermal properties of PLLA oligo-esters (M n(GPC) = 7800–39000) with only a small alkyl end group as dodecyl (C12H27) were nearly identical respect to nonend-capped PLLA.
Hypothetically, the effect of one end group can be most important on PLLA oligo-ester (M n(GPC) ~ 3500) that to polyester high molecular weights. However, the effect of linear alkyl end group (CH3−[CH2−CH2]m−CH2−) with systematic increase on PLLA oligo-esters has never been reported. In this work, a versatile synthesis of α-hydroxyl-ω-alkyl poly(L-lactide)s (PLLA)s is reported. In Scheme 1, different linear aliphatic alcohols (HO−CH2−[CH2−CH2]m−CH3, where m = 1, 2, 4, 5, 6, 7, 8, 9, and 10) has been successfully used as initiators/chain transfer agents in each new reaction from ROP of L-lactide (L-LA) catalyzed by Sn(Oct)2. The alkyl end group in PLLA was increased from butyl (C4H9) to docosyl (C22H45). α-Hydroxyl-ω-alkyl PLLA oligo-esters with M n(GPC) = 3520-4340 [M n(NMR) = 2120–2450] were obtained and characterized by 1 H and 13 C NMR, GPC, FT-IR and MALDI-TOF. The effects of the alkyl end group on PLLA oligo-esters were observed by DSC, TGA and SAXS.

Ring-opening polymerization (ROP) of L-lactide (LLA) catalyzed by tin(II) 2-ethylhexanoate Sn(Oct)2 in presence of primary aliphatic alcohols as initiators (CH3−[CH2−CH2]m−CH2−OH (ROH), where m = 1, 2,4,5,6,7,8,9, and 10)
Experimental
Materials
L-lactide (L-LA) [PURAC Biomaterials] was used without further purification. 1-butanol, 1-hexanol, 1-decanol, 1-dodecanol, 1-tetradecanol, 1-hexadecanol, 1-octadecanol, 1-eicosanol, 1-docosanol and tin(II) 2-ethylhexanoate [Sn(Oct)2] were purchased from Aldrich and used without further purification.
Typical procedure for the synthesis of α-hydroxyl-ω-docosyl poly(L-lactide) (PLLA10)
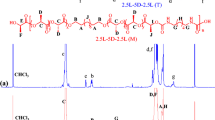
Polymerization was carried out in a 100 ml round-bottom flask previously dried. L-lactide (L-LA, 40 mmol, 5.760 g), 1 ml of a solution 0.123 M of tin(II) 2-ethylhexanoate Sn(Oct)2 in toluene and 1-docosanol (4 mmol, 1.306 g) were charged and heated to reflux by stirring them in an oil bath at 170 °C for 2 h (molar ratio L-LA/Sn(Oct)2 = 325 and L-LA/1-docosanol = 10). The oligo-ester obtained was precipitated from chloroform/methanol, recovered by filtration and dried under vacuum. The yield was calculated based on weight of monomer and initiator at the feed (yield = 69%), M n(NMR) = 2120, M n(GPC) = 3600, M w/M n = 1.24. Other PLLA oligo-esters (PLLA1-9) were prepared and isolated in the same manner as described in the PLLA10. Molecular weight was monitored by 1 H NMR. IR (cm-1): 2927 (νas, CH), 1757 (ν, C = O), 1457 (δs, CH), 1184 (νas, C−(C = O)−O), 1091 (νas, O−C−C). 1 H NMR data for α-hydroxyl-ω-docosyl poly(L-lactide) (PLLA10) (Fig. 1): (300 MHz, CDCl3, ppm) δ 5.18 (quartet, 1 H, [CH], PLLA), 4.37 (quartet, 1 H, [CH−OH], PLLA) 4.13 (two triplets, overlapping, 2 H, [CH 2O], docosyl), 1.60 (d, 3 H, [CH 3], PLLA), 1.52 (d, 3 H, [CH 3 ], PLLA), 1.50 (d, 3 H, [CH 3 ], PLLA), 1.24 (s, 2 H, [CH 2], docosyl), 0.87 (t, 3 H, [CH 3], docosyl). 13 C NMR data for α-hydroxyl-ω-hexyl poly(L-lactide) (PLLA2) (Fig. 2): (75 MHz, CDCl3, ppm) δ 174.99 (g), 170.04 (q), 169.49 (j), 69.23 (h), 68.91 (k), 66.58 (m), 65.58 (f), 31.22 (c), 28.31 (e), 25.28 (d), 22.38 (b), 20.40 (p), 16.73 (i), 16.54 (l), 13.87 (a).

1H NMR (300 MHz) spectrum in CDCl3 for the α-hydroxyl-ω-docosyl poly(L-lactide) [M n(NMR) = 2120 (PLLA10, Table 1)]

13C NMR (75 MHz) spectrum in CDCl3 for the α-hydroxyl-ω-hexyl poly(L-lactide) [M n(NMR) = 2260 (PLLA2, Table 1)]
Characterization
1 H and 13 C NMR spectra were recorded at room temperature on a Varian Unity Plus 300 (300 MHz 1 H and 75.47 MHz 13 C) and Bruker Avance 500 (500 MHz 1 H and 125 MHz 13 C). Chloroform-d (CDCl3) was used as solvent. Spectra were referenced to the residual solvent protons at δ 7.26 for CDCl3 in the 1 H NMR spectrum and the residual solvent carbons at δ 77.0 in the 13 C NMR spectrum. Matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) spectra were recorded in the linear mode by using a Voyager DE-PRO time-of-flight mass spectrometer (Applied Biosystems) equipped with a nitrogen laser emitting at 337 nm with a 3 ns pulse width and working in positive-ion mode and delayed extraction. A high acceleration voltage of 25 kV was employed. 2,5-Dihydroxybenzoic acid (DHB) at a concentration of 10 mg/ml in acetonitrile was used as matrix. Samples were dissolved in acetonitrile and mixed with the matrix at a molar ratio of approximately 1:100. FT-IR spectra were obtained with the attenuated total reflectance spectroscopy (ATR) technique on powder deposited over a diamond on zinc selenide (ZnSe) crystal in a Perkin-Elmer Precisely Spectrum 100 FT-IR spectrometer. GPC measurements were determined using a Waters gel permeation chromatograph equipped with a Waters 1515 Isocratic HPLC Pump and Waters 2414 refractive index (RI) detector. A set of two waters columns conditioned at 25 °C were used to elute samples at the flow rate of 1 ml/min HPLC-grade THF. Polystyrene standards (Polymer Laboratories) were used for calibration. Differential scanning calorimetry (DSC) was performed in a Mettler Toledo DSC822e instrument. Samples were sealed in aluminum pans. Two scans (25−180 °C and 0−180 °C) were performed with an initial heating rate of 10 °C/min and the fastest cooling rate of the instrument between runs under nitrogen purge. The melting points (T m) and crystallizations temperatures (T c) are given as the maximum and minimum of the endothermic and exothermic transition, respectively; and the data reported was taken from the second scan. The degree of crystallinity (x i) for the PLLA homopolymer was calculated from the endothermic peak area ΔH i by x i = ΔH i /ΔH i 0, where ΔH i 0 is the heat of fusion for perfect PLLA crystals (93 J/g) [11, 15]. A thermal analysis (TA) Q500 instrument was used for TGA. Samples with weights between 10 and 30 mg were scanned in Hi-Resolution mode with an initial heating rate of 10 °C/min under flux of nitrogen. SAXS measurements were taken at beamline BM16 at the European Synchrotron Radiation Facility (Grenoble, France). Samples were placed in between aluminum foils within a Linkam hot stage and heated at 10 °C/min while the SAXS spectra were recorded. Calibration of temperature gave a difference of approximately 7 °C between the temperature reading at the hot stage display and the real temperature at the sample. Long period (L) and lamellae thickness (D) were calculated by L = 2π/q max and D = L.x i, where q max and x i is the maximum scattering peak (by SAXS) and degree of crystallinity (by DSC), respectively.
Results and discussion
Synthesis and Characterization
Tin(II) 2-ethylhexanoate [Sn(Oct)2] have been previously reported as catalyst for ring-opening polymerization (ROP) of lactones as L-lactide (L-LA) [1–3]. Storey and coworkers investigated the ROP of ε-caprolactone (CL) and L-LA by Sn(Oct)2 [16] and stannous ethoxide Sn(OEt)2 [17] as catalysts, respectively, in the case of Sn(Oct)2 they found that 1-butanol is better initiator respect to ethylene glycol or 1,3-propanediol because a cyclic complex in the transfer reaction is not favored [16]. Usually, Sn(Oct)2 in presence of an alcohol is one of the best catalyst in the synthesis of poly(L-lactide) (PLLA) due to control of molecular weight and an appropriate route for the obtention of end-funtionalized poly(L-lactide) (PLLA). In our case, ROP of the L-LA was carried out with Sn(Oct)2 as catalyst and nine different aliphatic primary alcohols as initiators/chain transfer-agents (CH3−[CH2−CH2]m−CH2−OH (ROH), where m = 1,2,4,5,6,7,8,9, and 10) with a L-LA/Sn(Oct)2 and L-LA/ROH feed molar ratio of 325 and 10, respectively (Scheme 1). Moderate yields (62–71%) were achieved at 170 °C for 2 h. The value of theoretical degree of polymerization (DP) expected for all PLLA oligo-esters is 20. However, the DP obtained (by 1 H NMR) after purification is higher (DP(NMR) = 25–31). This result can be attributed to losses of oligomers in the recrystalization step. Nine different samples of poly(L-lactide) (PLLA1-10) were obtained by this route (Table 1). Unimodal distributions and moderate polydispersity (M w/M n = 1.20-1.33) were observed in all GPC chromatograms. Values of M n(GPC) are higher than M n(NMR). Overestimation of M n obtained by GPC for PLLA is a common feature, since polystyrene standards are used in the construction of the calibration curve, these different values were attributed to differences in hydrodynamic volume and molecular masses between polystyrene and PLLA [18, 19]. In the case of oligo-esters as poly(L-lactide) or poly(ε-caprolactone) M n values from the NMR analysis are close to the calculated theoretical M n values [18–22]. Therefore, the absolute M n was obtained from NMR analysis.
In Fig. 1, 1 H NMR spectrum from α-hydroxyl-ω-docosyl PLLA (PLLA10) is showed, where methyne g [–CH(CH3)–OH, δ 4.37, PLLA] and methyl a [CH 3 –, δ 0.87, docosyl] end groups are clearly seen. Two methyl end groups at δ 1.52 (e) and 1.50 (h) for PLLA10 also can be observed. Complementary, two triplets are overlapped at 4.13 ppm (c, docosyl) and were assigned to methylene end group adjacent to the ester group. Other peaks in the spectrum were assigned to different methynes [−CH(CH3)−O−] and methylenes [−CH 2 −] in the main chain oligo-ester and end group, respectively. The peaks assignation is consistent with the observed in other PLLA oligomers spectra [23, 24].
When 1-hexanol was used as initiator in the ROP of L-LA, 13 C NMR spectrum also shows evidence of the α-hydroxyl-ω-hexyl PLLA (PLLA6) end groups (Fig. 2). Terminal methyne close to hydroxyl m [−CH(CH3)−OH, δ 66.58] and methyl within the hexyl terminated a [CH3−(CH2)5−, δ 13.87] were observed. In carbonyl zone peaks three different signals can be distinguished: 1) ester carbonyl of the main chain j [−C(=O)−CH(CH3)−O−, δ 169.49], 2) ester carbonyl of the hydroxyl end group q [−C(=O)−CH(CH3)−OH, δ 170.04], and 3) ester carbonyl in the hexyl end group g [−CH(CH3)−C(=O)−O−(CH2)5−CH3, δ 174.99]. Peaks assignments for methynes and methyls were made comparing them with the 13 C NMR spectrum for PLLA reported by other authors [21, 24, 25]. Signals for methylenes were assigned using substituent additivity rules and literature data [26]. Carbon d appears with most difference between δ calculated and δ NMR (Table 2), d is a double γ-carbon with respect to methyl and ester group, and produces a shift to the right due to steric compression of a gauche interaction [26]. A similar phenomenon was observed for carbon gamma (γ) in the main chain for the PCL [19].
MALDI-TOF spectrum for α-hydroxyl-ω-docosyl (PLLA) (PLLA10) is showed in the Fig. 3a. Unimodal distribution similar to GPC is seen. A significant predominance of the linear PLLA10 oligo-ester was observed after analysis of signals. 1 H NMR confirmed this result. Tree type of signals can be observed in the view expansion of the zone between 1900 and 2125 uma (Fig. 3b), two were assigned to α-hydroxyl-ω-docosyl (PLLA) (PLLA10) doped with sodium (A) and potassium (B) ions and a third signal correspond to α-hydroxyl-ω-(carboxylic acid) PLLA (HA-PLLA) (C). The presence of HA-PLLA is the result of an initiation by water or linear dilactide (with OH groups) as impurity [23].

MALDI-TOF spectrum of the a α-hydroxyl-ω-docosyl poly(L-lactide) (PLLA10, Table 1). b Expanded view for the 1900-2125 m/z fragments
FT-IR spectrum of PLLA10 showed typical bands at 2927 (CH, ν), 1757 (C = O, ν), 1457 (CH, δ), 1184 (−C−(C = O)−O−, ν), and 1091 cm-1 (−O−C−C−, ν) [26]. Usually, the band for C = O stretching vibration (1757 cm-1) of PLLA is used as evidence of the formation of block copolymer in the poly(L-lactide-b-ε-caprolactone) [27, 28].
Thermal properties: DSC and TGA
Thermal properties of poly(L-lactide) (PLLA) oligo-esters (PLLA1-10) were examined by differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA). Effect of alkyl end group on thermal properties and crystalline nature of the PLLA was studied by DSC (Table 3). The thermal profile of α-hydroxyl-ω-butyl PLLA (PLLA1, Fig. 4) reveals characteristics of glass transition temperature (T g = 37 °C), crystallization temperature (T c1 = 78 °C), and melting temperature (T m1 = 141 °C); a similar pattern to PLLA is observed in other synthesized [25, 29] and commercials samples [29]. The values in melting point (T m = 137–142 °C) and enthalpy of fusion (ΔH m = 48–53 J/g) of PLLA samples are discrete, indicating similar crystallinity (x i) in all oligo-esters. Hence, the alkyl terminal group in all samples does not affect the crystallinity (x i ) of PLLA. Different result was found by Abayasinghe et al for poly(ethylene-b-L-lactide) (PE-b-PLLA) where the copolymer is more crystalline than homo-PLLA [11]. The effect of alkyl end group on T g is plotted in Fig. 5. T g gradually decreases with an increase in the percent of alkyl−CH2−[CH2−CH2]m−CH3 end group. The flexibility to PLLA is increased due to the attachment of a big alkyl end group as docosyl−CH2−[CH2−CH2]10−CH3. The contrary effect was reported by Frey and coworkers where the flexibility in hyperbranched polyglycerol was reduced by PLLA blocks [30].

DSC thermogram (second scan) of α-hydroxyl-ω-butyl poly(L-lactide) (PLLA1, Table 3)

Effect of alkyl end group on glass transition temperature (T g) of poly(L-lactide) (PLLA)
In Fig. 6, a new peak T C2 of cold crystallization exotherm for PLLA8-10 (11–15 wt % of alkyl end group) with respect to PLLA1 (Fig. 4) was observed. In based on comparison with T c1 of PLLA1 (Table 3, Fig. 4) the first peak at 79 °C was assigned to PLLA homopolymer environment. The second peak at 52 °C (T c2) was assigned to segments of PLLA [−CO−CH(CH3)−O−]n close to the alkyl end group (octadecyl to docosyl). In the case of α-hydroxyl-ω-docosyl PLLA (PLLA10) (Fig. 6c), where docosyl end group had 15% in weight, a phases separated can be seen with two melting points, the first slightly at 38 °C attributed to docosyl end group and the second at 138 °C assigned to PLLA. The second peak of cold crystallization exotherm (T c2) (Fig. 6c) appears because lower energy is necessary in the crystallization process for PLLA chains close to alkyl end group, due to docosyl melted. To corroborate this idea, tree new different samples with M n(NMR) = 1040–1910 were synthesized (Table 4). In Fig. 7, thermograms of PLLA with different M n and percent in weight end group (% EG) are showed. It is possible see as the second melting point peak (T m2 = 37–47 °C) is most intense as the % EG is higher (17–31%) and the T m1 attributed to PLLA decreased proportionally with the M n(NMR) from 130 to 121 °C. Finally, in the Fig. 7c a forceful segregation phases between docosyl (T m2 = 47) and PLLA (T m1 = 121) was observed. On the other hand, double cold crystallization (T c1 and T c2) profile can be seen in the Fig. 7a–b; however, the first peak of cold crystallization assigned to PLLA homopolymer environment (T c1 = 75–79 °C) was decreasing proportionally as the % EG has increased, even disappear with 31%. To our knowledge, the phenomenon of double cold crystallization has never been observed in previous works from PLLA [12] or PE-b-PLLA [10, 31, 32]. Our evidence shows that: 1) when alkyl end group (CH3−[CH2−CH2]m−CH2−) showed a percent in weight over 11% in the PLLA oligo-ester, a double cold crystallization was induced in the PLLA oligo-ester, 2) the double cold crystallization of PLLA is directly related with the segregation phase between alkyl end group and PLLA, and 3) Based on the presence of a double cold crystallization in the thermograms is possible to affirm than that start segregation phases process is at % EG ≥ 11 for PLLA oligo-esters with M n(NMR) = 2120–2450.

DSC thermograms (second scan) of a α-hydroxyl-ω-octadecyl poly(L-lactide) (PLLA8), b α-hydroxyl-ω-eicosyl poly(L-lactide) (PLLA9) and c α-hydroxyl-ω-docosyl poly(L-lactide) (PLLA10, Table 3)

DSC thermograms (second scan) from α-hydroxyl-ω-docosyl poly(L-lactide) with different percent of end group (%EG) and M n(NMR): a 1910, b 1320 and c 1040
In the α-hydroxyl-ω-alkyl PLLA oligo-esters is expected that alkyl end group have an effect on thermal stability, and by thermogravimetric analysis (TGA) was observed, for example: Jamshidi [33] reported an increase in degradation temperature [detected by TGA] of the α-acetyl-ω-(carboxyl acid) PLLA (PLLA acetylated) respect to α-hydroxyl-ω-(carboxyl acid) PLLA. In our samples (Fig. 8), thermogravimetric analysis (TGA) of PLLA oligo-esters showed only one inflection temperature for ω-butyl and ω-docosyl end group at 215 and 223 °C, respectively. The difference in thermal degradation probably is due to steric hindrance of the end group (docosyl respect to butyl) toward intermolecular and intramolecular transesterification in the formation of monomer and cyclic oligomers.

Thermal degradation of PLLA oligo-esters with different terminal group: a α-hydroxyl-ω-butyl and b α-hydroxyl-ω-docosyl
Small-angle x-ray scattering (SAXS)
It is well-known that PLLA is a semicrystalline polyester with lamellar structure in its crystalline phase that can be detected by small-angle x-ray scattering (SAXS). With this idea, in the Fig. 9 the SAXS curves for α-hydroxyl-ω-butyl poly(L-lactide) (PLLA 1) and α-hydroxyl-ω-docosyl poly(L-lactide) (PLLA10) at different temperature are showed. In Fig. 9a, scattering of PLLA1 at lower temperature (27 °C) is not seen. Typically, scattering of PLLA commercial samples at room temperature has not been observed. However, when increasing temperature one scattering peak at T = 80 °C and q max = 0.54 nm-1 is observed just after the beginning of the crystallization T c1, based on q max the long period (L = 2π/q max) was calculated, L = 11.6 nm, indicating the distance center-to-center between lamellae. When the temperature was gradually increasing the maximum peak (q max) is shifted toward higher value of L. Thus, lamellae growth in PLLA is proportional to the increase in L. Reaching a maximum peak at T = 133 °C with q max = 0.46 nm-1 and L = 13.6 nm after its crystallization temperature (T c1 = 78 °C) and below the melting temperature (T m = 141 °C); with higher temperature (T > T m) the peak disappears totally when PLLA was melted and scattering is not seen. Therefore, any order after PLLA melting point is not observed. In the case of PLLA10 (Fig. 9b) respect to PLLA1, one peak at 27 °C with q max = 1.01 nm-1 (L = 6.22 nm) is observed. Scattering at 27 °C is induced by docosyl end group due to segregation phases between oligo-ester and aliphatic terminal group. Similar patron was observed with octadecyl end group. When the temperature was raised, a maximum peak a q max = 0.63 nm-1 (T = 116 °C) is observed. With this result, the maximum value of L for PLLA10 is 9.9 nm and respect to 13.6 nm of PLLA1, a decrease is seen. Based on L and crystallinity fraction (by DSC), the lamellae thickness (D) in both samples was calculated and a minor lamellae thickness in PLLA10 (D = 5.0 nm) respect to PLLA1 (D = 7.3 nm) was obtained. This result is explained in terms of the steric hindrance of docosyl end group toward lamellar growth.

SAXS curves (Iq2 vs q) plotted during heating for: a α-hydroxyl-ω-butyl PLLA and b α-hydroxyl-ω-docosyl PLLA
Conclusion
Poly(L-lactide) (PLLA) oligo-esters were synthesized by ring-opening polymerization (ROP) of L-lactide (L-LA) catalyzed by tin(II) 2-ethylhexanoate Sn(Oct)2 and using aliphatic alcohols CH3−[CH2−CH2]m−CH2−OH as initiators with a systematic sequence in the increase (m =1–10). Monofuntionality α-hydroxyl-ω-alkyl PLLA were found in all oligo-esters characterized. PLLA oligo-esters showed evidence of the effects of alkyl CH3−[CH2−CH2]m−CH2− end group on their thermal properties detected by DSC and TGA. Glass transition temperature (T g) is gradually decreased with an increase in the percent of alkyl−CH2−[CH2−CH2]m−CH3 end group, as results alkyl end group provides more flexibility to PLLA. The effect of the alkyl end group on crystallinity of PLLA was discrete. An important effect of alkyl end group on PLLA was observed with a double cold-crystallization (T c) at higher values of percent in weight of end-group (≥ 11%) and directly related with the segregation phases between alkyl end group and PLLA. TGA analysis revealed that PLLA oligo-esters are more thermally stable with docosyl (−C22H45) respect to butyl (−C4H9) end group, probably is due to steric hindrance of the end group (docosyl respect to butyl) toward intermolecular and intramolecular transesterification. Lamellar growth in PLLA was sterically hindered by docosyl (−CH2−[CH2−CH2]10−CH3) end-group with a decreasing value in the lamellae thickness (D).
References
Fukushima K, Kimura Y (2006) Polym Int 55:626–642
Stolt M, Krasowska K, Rutkowska M, Janik H, Rosling A, Södergård A (2005) Polym Int 54:362–368
Korhonen H, Helminen A, Seppälä JV (2001) Polymer 42:7541–7549
Gottschalk C, Frey H (2006) Macromolecules 39:1719–1723
Miola-Delaite C, Hamaide T, Spitz R (1999) Macromol Chem Phys 200:1771–1778
Kowalski A, Duda A, Penczek S (2000) Macromolecules 33:689–695
Fan Y, Chen G, Tanaka J, Tateishi T (2005) Biomacromolecules 6:3051–3056
Spasova M, Mespouille L, Coulembier O, Paneva D, Manolova N, Rashkov L, Dubois P (2009) Biomacromolecules 10:1217–1223
Karanikolopoulos N, Zamurovic M, Pitsikalis M, Hadjichristidis N (2010) Biomacromolecules 11:430–438
Wang Y, Hillmyer MA (2001) J Polym Sci Part A Polym Chem 39:2755–2766
Abayasinghe NK, Glaser S, Prasanna K, Perera U, DWJr S (2005) J Polym Sci Part A Polym Chem 43:5257–5266
Kurokawa K, Yamashita K, Doi Y, Abe H (2006) Polym Degrad Stab 91:1300–1310
Kurokawa K, Yamashita K, Doi Y, Abe H (2008) Biomacromolecules 9:1071–1078
Kobori Y, Iwata T, Doi Y, Abe H (2004) Biomacromolecules 5:530–536
Ouchi T, Ohya Y (2004) J Polym Sci Part A Polym Chem 42:453–462
Storey RF, Sherman JW (2002) Macromolecules 35:1504–1512
Storey RF, Mullen BD, Desai GS, Sherman JW, Tang CN (2002) J Polymer Sciences. Part A: Polymer Chemistry 40:3434–3442
Huang C-H, Wang F-C, Ko B-T, Yu T-L, Lin C-C (2001) Macromolecules 34:356–361
Báez JE, Martínez-Richa A, Marcos-Fernández A (2005) Macromolecules 38:1599–1608
Báez JE, Martínez-Rosales M, Martínez-Richa A (2003) Polymer 44:6767–6772
Finne A, Albertsson A-C (2004) J Polym Sci Part A Polym Chem 42:444–452
Chen H-L, Ko B-T, Huang B-H, Lin C-C (2001) Organometallics 20:5076–5083
Kricheldorf HR, Hachmann-Thiessen H, Schwarz G (2004) Biomacromolecules 5:492–496
Lemmouchi Y, Perry MC, Amass AJ, Chakraborty K, Schué F (2007) J Polym Sci Part A Polym Chem 45:2235–2245
Messman JM, Scheuer AD, Storey RF (2005) Polymer 46:3628–3638
Silverstein RM, Webster FX, Kiemle DJ (2005) Spectrometric identification of organic compounds. John Wiley & Sons, New Jersey
Qian H, Bei J, Wang S (2000) Polym Degrad Stab 68:423–429
Huang M-H, Li S, Coudane J, Vert M (2003) Macromol Chem Phys 204:1994–2001
Takizawa K, Nulwala H, Hu J, Yoshinaga K, Hawker CJ (2008) J Polym Sci. Part A: Polym Chem 46:5977–5990
Gottschalk C, Wolf F, Frey H (2007) Macromol Chem Phys 208:1657–1665
Castillo RV, Müller AJ, Lin M-C, Chen H-L, Jeng U-S, Hillmyer MA (2008) Macromolecules 41:6154–6164
Ring JO, Thomann R, Mülhaupt R, Raquez J-M, Degée P, Dubois P (2007) Macromol. Chem Phys 208:896–902
Jamshidi K, Hyon S-H, Ikada Y (1988) Polymer 29:2229–2234
Acknowledgements
J.E.B. is much indebted to the Instituto de Ciencia y Tecnología de Polímeros (CSIC), Consejo Nacional de Ciencia y Tecnología (CONACYT, México) and Sistema Nacional de Investigadores (SNI, México). J.E.B. and A.M.F thanks to PURAC Biomaterials for the donation of L-lactide (L-LA) monomer. A.M.F. and J.E.B. thanks the Ministerio de Educación y Ciencia for its financial support in accessing the Synchrotron, and François Fauth and Ana Pastor for their help on the beamline BM16 (Grenoble, France). J.E.B. thanks to Jesús L. Pablos and Mario Luzón for obtaining GPC chromatograms.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Báez, J.E., Marcos-Fernández, Á. & Galindo-Iranzo, P. Exploring the effect of alkyl end group on poly(L-lactide) oligo-esters. Synthesis and characterization. J Polym Res 18, 1137–1146 (2011). https://doi.org/10.1007/s10965-010-9517-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10965-010-9517-y