
Abstract
Cerium incorporated MCM-22 (Ce-MCM-22) was successfully synthesized by hydrothermal method. Incorporation of Ce is achieved by the optimization of method for synthesis by varying (i) Concentration of sructure directing agent or template, (ii) Silica to ceria ratio and (iii) Crystallization time. Change in phases during crystallization, variation of physico-chemical properties with respect to gel composition and framework substitution of cerium are evidenced by XRD, UV–Vis DRS, SEM, N2 adsorption, FT-IR, Ammonia TPD and solid-state MAS-NMR characterisations. It is found that incorporation of Ce into the zeolite frame work has successfully occurred by simple method of optimisimg molar gel composition. Interestingly, prepared samples of Ce-MCM-22 have also been found to be more effective catalysts for the removal of trace olefins from aromatic streams.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Zeolites have been found to be valuable catalysts in petrochemical and fine chemical processes because of their unique shape selectivity and catalytic activity [1,2,3,4]. Zeolites are the solid acid catalysts mainly used in acid catalysed reactions, but their catalytic functionality can be varied through the incorporation of hetero atoms into the frame work. The incorporation of different metals into the zeolite framework can deliver heteroatomic zeolite materials with modified physico- chemical properties thus can lead to remarkable catalytic activity. Transition and inner transition metals such as Ti, Fe, Zr, Nb, Ce, La etc., have been introduced into the framework of different zeolitic structure thus making these materials has highly active catalysts for their application in new chemical processes [5,6,7,8,9,10]. These framework heteroatoms may generate special acid sites that shows synergistic effect on the acid catalyzed reactions such as alkylation, thus can lead to an increased efficiency of the catalyst. Also, the strength of the acid sites can be fine-tuned by modifying the type and the number of tetrahedral atoms in the zeolite frame work. MWW zeolites are important class of materials because of the possibility of diverse structural, textural and compositional modifications [11]. MCM-22 was the first [12, 13] invented MWW zeolite that contains two independent pore systems: one consists of two-dimensional sinusoidal 10-ring channels, and the other large 12-ring supercages connected by 10-ring windows. Furthermore, its external surface consists of 12-ring cups. Because the acid sites in those pockets are accessible to large organic molecules, MCM-22 has shown to be applicable to a wide variety of chemical reactions [14,15,16]. Modification of MWW zeolites with lanthanide ions may give an opportunity to obtain zeolite catalysts with better performance. However, a feasible method for the preparation of framework-substituted lanthanide zeolites remains a big challenge because of size difference between lanthanides and the Si4+ ion [17] that has to be substituted in the framework. For the preparation of heteroatom incorporated zeolites, different methods such as conventional hydrothermal crystallization, dry-gel synthesis, fluoride modification, acid hydrolysis and microwave-assisted methods have been reported [18,19,20,21,22,23,24]. Although highly active zeolite catalysts can be obtained using various synthesis strategies, most of these routes are complicated and time-consuming resulting into difficulty in reproducibility and feasibility for large-scale synthesis. Moreover, these methods of synthesis takes place in more than one step which are very sensitive to method of preparation and type of precursor sources for silica and alumina. In continuation of our earlier work [25] for synthesising MCM-22, in the present study we have successfully synthesised frame work incorporated Ce-MCM-22 by optimising the experimental conditions and gel composition. Unlike the reported synthesis procedures which take 6–7 days for crystallisation, here we reported a short crystallisation time of 48 h for the synthesis of Ce-MCM-22. In addition to this the amount of organic template used is half of to that of reported in the literature [23, 24], which is the most expensive rawmaterial used in zeolite preparation. Therefore we claim it is an economically viable synthesis procedure as compared to reported procedures [23, 24]. Catalytic activity of the prepared catalysts was tested for olefin removal from aromatic streams, which is technically called as aromatics purification in petrochemical industry [26, 27]. Olefinic contaminants will have adverse effect on the donstream seperation processes, therefore it has to be removed before they route to other processes. Based on our experience in the area of zeolite catalysis in petrochemical processes, we revealed that heteroatom zeolites can become a good catalyst for such processes.
2 Experimental
As reported in the literature [28], synthesis mixtures with SiO2/Al2O3 = 30, H2O/SiO2 = 15, Na/SiO2 = 1.5 and HMI/SiO2 = 0.2 were prepared using Colloidal silica (40 wt% SiO2, LUDOX AS-40, Sigma-Aldrich), Sodium aluminate powder (50–55% Al2O3, 40–45% Na2O, Sigma-Aldrich), Sodium hydroxide (SD fine chemicals Ltd), Hexamethyleneimine (99%, Aldrich) as a template or structure directing agent (SDA) and deionised water. In a typical synthesis procedure, NaOH was added to the deionised water followed by the slow addition of NaAlO2 under vigorous stirring. To this solution, hexamethylene imine (HMI) was added followed by the drop wise addition of colloidal silica under vigorous stirring. Then the mixture was stirred continuously until it forms a homogeneous gel. The resulting gel was transferred into a teflon lined autoclave and then subjected to hydrothermal treatment at 443 K under stirring at 250 rpm for 24–72 h (sample 1 to 3 in Table 2). For Ce-MCM-22, Cerium (III) nitrate hexahydrate (Sigma Aldrich) is used as cerium precursor. Composition of synthesis mixture was SiO2/Al2O3 = 38–43, H2O/SiO2 = 15, Na/SiO2 = 1.5, HMI/SiO2 = 0.2–0.3 and SiO2/CeO2 = 100–150 (sample 4 to 9 in Table 2). For synthesis of Ce-MCM-22, requisite amount of Ce precursor solution was added to the mixture of sodium aluminate and sodium hydroxide solution followed by the addition of hexamethyleneimine and colloidal silica. The resulting gel was transferred into a teflon lined autoclave and then subjected to hydrothermal treatment at 443 K under stirring at 250 rpm for 48–72 h (sample 4 to 9 in Table 2). The resultant products were filtered, thoroughly washed with deionized water, and dried at 393 K. All the samples were calcined at 823 K for 6 h in air to remove the SDA. The hydrogen forms of the samples were obtained by exchanging the calcined material with NH+4 using 1 M NH4NO3 aqueous solution followed by calcination in air at 823 K for 6 h.
2.1 Shaping of powders into extrudates
For catalytic performance evaluation, both Ce-MCM-22 and MCM-22 zeolite powders were shaped in the form of 1.5 mm cylindrical extrudates using alumina as a binder (zeolite to binder ratio was maintained as 70:30) and calcined in air at 823 K for 6 h.
2.2 Characterisation
Characterization techniques used to elucidate the textural and structural properties of the prepared zeolite materials were XRD, ICP, N2 adsorption, 29Si and 27Al MAS NMR, UV–Vis DRS, FTIR, Ammonia TPD, and SEM. XRD patterns were collected on the Bruker D8 Advance powder diffractometer using Ni-filtered Cu Kα radiation source at 40 kV and 20 mA, from 2 to 50° with a scan rate of 2°/min. BET surface area and pore size were measured at 77.2 K using a Micrometrics 3Flex Surface Characterization Analyser. The samples were degassed at 673 K to a vacuum of 10–3 Torr, before analysis. MAS NMR of the calcined samples were measured by Bruker-Advance III-HD. ICP of the synthesised samples were measured by using ICAP-7600 Thermo Fisher instrument. IR spectra of samples were obtained by using Thermo Fisher IS50, ATR mode. UV–Vis DRS spectra were measured with spectrometer Agilent Technology, Cary-5000 UV–Vis NIR. BaSO4 was used as an internal standard for recording of UV–Vis DRS spectra. Ammonia TPD was measured for total acidity of the zeolite on Autochem 2920. Scanning electron microscopy (SEM) was performed using Nova Nanosem-650 in high-vacuum mode at low voltage.
2.3 Catalytic activity test
Experimental raw feedstock with a bromine index (BI) of 650 mg/100 gm was an aromatic intermediate product was obtained from a commercial catalytic reforming unit of Reliance Petrochemical complex, Reliance Industries Ltd. The main components of this aromatic feedstock were C8 to C10 aromatics, the composition of which is presented in Table 1. Olefins concentraion in feed is represented as bromine index.
5 g of extrudates were added to 35 g of aromatic feed stock in a stainless-steel bomb of 70 ml capacity. The reactor was purged with nitrogen to remove air and was closed. The bomb reactor was heated at 448 K temperature for the duration of 3 h. After this, the reactor was cooled to ambient conditions. The hydrocarbon liquid was separated from the solid catalyst and was examined for the concentration of olefins.
2.4 Product analysis
Bromine Index method (ASTM D-1491) was used to determine the olefin content in aromatic hydrocarbon. Specifically, the Bromine Index (BI) is defined as the number of milligrams of bromine consumed by 100 g of hydrocarbon sample. The number of milligrams of bromine absorbed by 100 g of a hydrocarbon or a hydrocarbon mixture indicates the percentage of double bonds present. A measure of the reduction in Bromine Index of the product from the process represents the degree of olefin conversion. Olefins conversion was calculated as follows:
Y = Percentage of olefins conversion or reduction in bromine Index.
BI0 = The bromine index of the reactant feed.
BI = The bromine index of the product stream.
3 Results and discussion
3.1 Effect of crystallization time on the synthesis of MCM-22
Hydrothermal treatment for different time durations such as 24, 48 and 72 h was carried out to find the effect of crystallization time on the physico-chemical properties. The molar gel compositions for the first set of three samples 1, 2 and 3 are similar which are presented in Table 2. XRD patterns of the as-synthesised samples are shown in Fig. 1. By comparing the peak positions and intensities of the XRD patterns with those reported in earlier studies [29], XRD phases in sample 1 and sample 2 with crystallisation time of 24 h and 48 h are observed with pure phases of MCM-22. Whereas, XRD patterns of the sample 3 with crystallisation time of 72 h shows an additional peak at 2θ = 9.45 and 22.4°. These additional peaks are identified as phase of FER (ferrierite, peak shown as F in XRD pattern). This indicates that longer crystallisation time under present synthesis conditions gives a mixture of MCM-22 and FER phase and not favourable for the formation of pure MCM-22.

XRD patterns of as-synthesised samples: (a) sample-1, (b) sample-2 and (c) sample-3
3.2 Effect of crystallisation time on the incorporation of Ce in MCM-22
In view of optimised conditions developed for synthesis of MCM-22, attempts were made to study the effect of crystallisation time on the synthesis of Ce-MCM-22 (Table 2) with the molar gel composition of SiO2/Al2O3 = 38, CeO2/SiO2 = 150, H2O/SiO2 = 15, Na/SiO2 = 1.5 and HMI/SiO2 = 0.2. As shown in Fig. 2, XRD patterns of sample 4 with 48 h of crystallisation time resulted in amorphous phase, subsequently, sample 5 is prepared with same gel composition by increasing the crystallisation time to 72 h. XRD patterns showed the characteristic peaks of Ce-MCM-22 along FER impurity. This shows that longer crystallisation times are not suitable for the incorporation of Ce in to the frame work of MCM-22. Similar observations were made in the synthesis of MCM-22 also. Here we couldn’t achieve the phase purity of Ce-MCM-22 while changing the crystallisation time, therefore we made an attempt to see the template effect on the phase purity of zeolite in the next case.

XRD patterns of as-synthesised samples: (a) sample-4, (b) sample-5, (c) sample-6 and (d) sample-7
3.3 Effect of template concentration on the incorporation of Ce in MCM-22
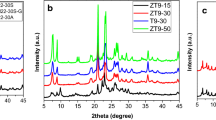
As stated in the earlier reports [30], template concentration plays a key role in the rate of nucleation as well as crystallisation of zeolites, we made an attempt to synthesise pure form of Ce-MCM-22 by varying the template concentration. Accordingly, sample 6 (shown in Table 2) was synthesised with slightly higher template concentration, HMI/SiO2 = 0.23 under an optimised crystallisation time 48 h, same as in case of MCM-22. This resulted in the formation of Ce-MCM-22 phase (Fig. 2) along with a small impurity of FER. As we observed improvement in the phase purity by increasing template concentraion, sample 7 was prepared with further increase in template concentration (HMI/SiO2 = 0.26), where in the final molar gel composition is SiO2/Al2O3 = 38, SiO2/CeO2 = 150, H2O/SiO2 = 15, Na/SiO2 = 1.5 and HMI/SiO2 = 0.26. XRD pattern (Fig. 2) of sample 7 is observed with pure phase of Ce-MCM-22 without any FER phase, which suggests that increased template concentration in the gel can promote the incorporation of Ce into the framework. Similarly, when the same conditions are applied to synthesise Ce-MCM-22 (sample 8 in Table 2) with high ceria content wherein gel composition is SiO2/Al2O3 = 43, SiO2/CeO2 = 100, H2O/SiO2 = 15, Na/SiO2 = 1.5 and HMI/SiO2 = 0.26, XRD patterns (Fig. 3) of sample 8 was observed with Ce-MCM-22 and FER phases. Subsequently to avoid the formation of impurities, sample 9 was prepared with the increased template concentration (HMI/SiO2 = 0.3), wherein the molar gel composition is SiO2/Al2O3 = 43, CeO2/SiO2 = 100, H2O/SiO2 = 15, Na/SiO2 = 1.5 and HMI/SiO2 = 0.3. As depicted from the XRD patterns in Fig. 3, this resulted in pure phase of Ce-MCM-22 with negligable amount of FER phase. These observations clearly shows that optimised crystallisation conditions and template concentration restricts the formation of impure phases thus forming pure Ce-MCM-22. It is important to note that though the template concentrations are increased to achieve phase purity, it is very less compared to literature reports. Therefore, we can infer that the amount of template used is in the present investigation is (HMI/SiO2 = 0.3) is half of to that reported [24] in the literature (HMI/SiO2 = 0.6).

XRD patterns of as-synthesised samples: (a) sample-8 and (b) sample-9
3.4 Powder X ray diffraction studies
In the present investigation, Sample 2, sample 7 and sample 9 are observed with high phase purity, XRD profiles of assynthesised and calcined samples are shown in Fig. 4, one distinct difference between the assynthesised and calcined samples is detected in the range of 2θ = 12–25°, wherein the X-ray patterns of calcined sample are characterized by sharp and separated peaks. Another region that distinguishes as-synthesised from calcined material occurs in the range between 2θ = 26° and 29° [31, 32]. Here as-synthesised sample presents only two peaks observed at 2θ = 26.07° and 26.60°, while the calcined sample shows four sharp peaks at 2θ = 26.23°, 27.14°, 28.08° and 28.84°. The percentage of crystallinity shown in Table 2 was assessed by comparing the sum of intensities of the peaks appearing at 2θ = 7–10 and 24–26° in the calcined samples to those same intensities found in the fully crystalline reference material. Percentage crystallinity of MCM-22 (sample 2) is higher than that of Ce-MCM-22 (sample 7 and 9). Subsequently, with increasing the Ce loadings crystallinity is further decreased, this may be because the insertion of Ce in to the framework slows down the crytallisation rate. In samples of Ce-MCM-22 (sample 7 and 9), the CeO2 quantity is about 1.7 and 2.8 wt% to the total amount of the tetrahedral oxides. There are no charecteristic peaks of CeO2 are observed even with 2.8 wt% CeO2 used to the total weight of zeolite used in the synthesis. This indicates that Ce could have inserted into the frame work of zeolite or a part of CeO2 may highly dispersed in the pores of the zeolite which is out of XRD detection limits.

XRD patterns of calcined samples: (a) sample-2, (b) sample-7 and (c) sample-9
3.5 Thermogravimetric analysis
The weight loss on ignition (LOI) is determined by thermogravimetric analysis. LOI of each sample varies based on the amount of template used in the synthesis as well as efficacy of washing carried out after the synthesis, 15 to 20% of weight loss is observed on the samples prepared. In the TGA of MCM-22 and Ce-MCM-22, the initial weight loss up to 453 K was due to desorption of physically adsorbed water, while the weight loss at 473–923 K is due to the decomposition of hexamethyleneimine used as template in the synthesis.
3.6 UV–Vis diffuse reflectance spectra
We have carried out the UV–Vis DRS analysis of selected calcined samples (sample 2, 7 and 9) which are obtained with pure phases in synthesis. Based on the wavelength of the bands observed, the oxidation state and chemical environment of metal atoms can be identified. As shown in Fig. 5, DRS spectra of MCM-22 (sample 2) doesn’t show any bands, while Ce-MCM-22 samples (sample 7 and 9) shows a broad band in the range of 230–300 nm. A well defined absorption band at 260 nm and less intense shoulder at 290 nm are attributed to Ce3+ and Ce4+ respectively, in tetrahedral coordination, indicating the incorporation of Ce into the framework of zeolite [23, 24, 33]. Although there are strong peaks assigned to the frame work Ce, it is possible that both intra-framework and extra-framework cerium species are present because of the broadness of the large absorption band [34].

UV–Vis DRS spectra of calcined samples: (a) sample-2, (b) sample-7 and (c) sample-9
3.7 Ammonia TPD
The acidity of the MCM-22 (sample 2) and Ce-MCM-22 (sample 7 and 9) was determined from temperature programmed desorption of ammonia (NH3-TPD). As shown in Fig. 6, all these samples exhibit two well resolved desorption peaks: the low-temperature peak (LTP) at 453-473 K and the high-temperature peak (HTP) at 573-723 K. Generally, LTP and HTP correspond to weak and strong acid sites, respectively. We couldn’t see much difference in the ratio of weak to strong acid ammonia desorption peak areas in MCM-22 and Ce-MCM-22 samples indicating that amount of strong and weak acid sites remains almost same in all the samples. Total acidity of sample 2, sample 7 and sample 9 is 1.23, 1.1 and 1. 15 mmol/g.cat., respectively.

Ammonia TPD measurements of calcined samples: (a) sample-2, (b) sample-7 and (c) sample-9
3.8 N2 adsorption and desorption isotherms
Nitrogen adsorption isotherms of calcined materials are compared in Fig. 7. The isotherm of MCM-22 (sample 2) was type I due to the microporous nature of the material, while that Ce-MCM-22 samples (sample 7 and 9) showed type IV isotherm with a hysteresis loop at p/po = 0.4 for capillary condensation, which indicates the successful formation of mesopores after the incorporation of Ce in MCM-22. BET surface area of MCM-22 was 480 m2/g, with pore volume of 0.4 cm3/g, while the BET surface area of Ce-MCM-22 (sample 7) was 470 m2/g with mesopore volume of 0.47 cm3/g indicating that mesopores are dominant. Sample 9, with high Ce content has observed with slightly low surface area (445 m2/g) may because of low crystallinaity. However, this sample is showing high pore volume of 0.51 cm3/g, because of high Ce incorporation into the frame work.

N2 adsorption isotherms of calcined samples: (a) sample-2, (b) sample-7 and (c) sample-9
3.9 29Si and 27Al MAS NMR spectra
29Si magic angle spinning (MAS) NMR were measured to determine the chemical environment of silica or to count the number of crystallographic sites in purely siliceous materials, and the 27Al MAS NMR to investigate the framework and extra framework species were used. The NMR spectra are sensitive to range of local interactions which provide detailed spatial and chemical information. 29Si NMR has shown to be sensitive to the substitution of range of other heteroatoms into the frame work and can be often be used to prove that these ions have been substituted. 29Si chemical shift is significantly shifted by the substitution of heteroatom in the frame work [35]. Figure 8 shows the 29Si-MAS NMR spectra of sample 2, sample 7 and sample 9.

29Si-MAS NMR spectra of calcined samples: (a) sample-2, (b) sample-7 and (c) sample-9

27Al-MAS NMR spectra of calcined samples: (a) sample-2, (b) sample-7 and (c) sample-9
The chemical shift in the range of −107 to −117 ppm are attributed to Si (0Al) and are denoted as Q4 sites in the spectra. The chemical shift at −103 ppm corresponds to Si (1Al) which are also called as Q4 sites. In addition to this, there was a noticeable 29Si resonance at −96 pmm (Q3 sites), this peaks is attributable to Si(OH) groups suggesting that MWW layers has larger number of surface hydroxyl groups [36]. The peaks of Ce-MCM-22 were shifted to up field relative to those of MCM-22, suggesting the incorporation of Ce ion into the frame work. The chemical shift toward the up field is more significant in sample-9 where the Ce content is higher compared to the sample-7 with low Ce content.
As shown in the Fig. 9, 27Al MAS NMR spectra of all the samples showed one signal at ca.50 ppm, indicating that Al atoms were tetrahedral coordinated in the zeolite framework, apart from this there is a peak visible around 0 ppm, indicating the presence of small quantity of octahedral coordinated aluminium in all the samples.
3.9.1 FTIR
As shown in Fig. 10, FT-IR bands observed in the range 820–850 cm−1 are assigned to O–Si–O symmetrical stretch. The bands in the range 1225–1250 cm−1 correspond to the external symmetric T–O–T stretching (where T = Si or Al) and the bands in the range 1030–1050 cm−1 are assigned to internal asymmetric T–O–T stretching, which is also in agreement with earlier studies [37, 38]. The bands in the range 400–450 cm−1 are assigned to T–O bending and the bands at 594 and 550 cm−1 are attributed to the presence of double 6 MR in the MCM-22 [39, 40]. The positions of bands due to vibrations of external linkages are very sensitive to structure. It is clearly seen from the FT-IR spectra that IR band around 1086 cm−1 for T–O bonds in TO4 tetrahedra of pure MCM-22 (sample 2) shifted slightly to lower range of 1081 cm−1 for sample 7 and sample 9 (Ce-MCM-22), thus suggesting the incorporation of Ce in the frame work [23, 41].

FTIR spectra of calcined materials: (a) sample-2, (b) sample-7 and (c) sample-9
3.9.2 Scanning electron microscopy
SEM images of sample 2, 7 and 9 are shown in Fig. 11. The primary crystals of MCM-22 (sample 2) show the well-known platelet morphology of MWW zeolite. As can be seen from SEM images, the particle size of the platelets are < 1 µm. From observed images it is clearly shows that particle size and morphology of Ce-MCM-22 and MCM-22 are nearly similar.

SEM images of as-synthesised: a sample-2, b sample-7 and c sample-9
Ce.
4 Catalytic performance
Catalytic performance of the MCM-22 and Ce-MCM-22 was evaluated for the conversion of olefins from a commercial C8 + aromatics stream. It is well known that the alkylation of aromatics with olefins over solid acid catalyst proceeds via the generally accepted carbenium ion mechanism illustrated in Scheme 1. On the basis of this mechanism, the olefin molecules adsorbed on the surface of the zeolite was attacked by the Bronsted acid or Lewis acid sites thus resulting in the reduction of double bond to form a carbenium ion. The electrophilic attack of the carbenium ion on the aromatic π-electrons leads to alkylation reaction thus resulting in alky benzenes and polyalkyl benzenes [42, 43].

Alkylation of aromatics by olefins over zeolite surface
We explored the olefin conversion activity of zeolites obtained with pure phases, which are, sample 2 (MCM-22), sample 7 and 9 (Ce-MCM-22). As can be seen in Fig. 12, olefin conversion acivity over Ce-MCM-22 samples was significantly higher than that of MCM-22, trend of olefin conversion activity is as follows: Ce-MCM-22 (SiO2/CeO2 = 100) > Ce-MCM-22 (SiO2/CeO2 = 150) > MCM-22. Olefin coversion over MCM-22 is 29% while that in Ce-MCM-22 (SiO2/CeO2 = 150) and Ce-MCM-22 (SiO2/CeO2 = 150) is 53 and 63% respectively. It could be noted that, as the amount of Ce content increases catalytic activity increases. Ce+4 present in the frame work with out bringing about local charge imbalance generates Lewis acid sites and makes the catalyst more active in olefin conversion. Substitution of lattice silica of zeolites by various metals and there enhanced catalytic performance were already reported in the literature [44, 45]. In addition to this there is also possibility of small amount of CeO2 species that are present outside the frame work on MWW layers, which also acts a lewis acid sites. Therefore, it is understood that Ce is playing a vital role enhancing the catalytic activity.

Olefin conversion acitivity of: sample-2 (MCM-22); sample-7 (Ce-MCM-22, SiO2/CeO2 = 150); sample 9 (Ce-MCM-22, SiO2/CeO2 = 100)
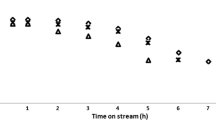
In order to see the leaching effect of Ce from the frame work of zeolite, we also carried out the regeneration studies of sample 9 (Ce-MCM-22, SiO2/CeO2 = 100) with high loadings of Ce content. Samples after each catalytic run was filtered, oven dried at 393 K for 12 h, followed by calcination in air at 823 K for 8 h. From Fig. 13, it is observed that even after 5 cycles of regeneration olefin conversion remains same. This clearly indicates that there is no leaching of Ce from the frame work of zeolite and structure remains intact.

Olefin conversion acitivity of fresh and regenerated sample: sample-9 (Ce-MCM-22, SiO2/CeO2 = 100)
5 Conclusions
Framework-substituted Ce-MCM-22 has been synthesized by hydrothermal synthesis while optimizing gel composition and experimental conditions. Ce-MCM-22 zeolites were prepared with varying amounts of Ce in the framework. Template concentration has played a vital role in controlling the impure phases. Incorporation of Ce in the frame work is evidenced by the 29Si and 27Al MAS NMR, FTIR and UV–Vis DRS techniques. Ce incorporated MCM-22 was observed with high catalytic activity towards removal of olefins from the C8+ aromatic stream to produce olefin free C8+ aromatics stream. Ce+4 present in the frame work with out bringing about local charge imbalance generates Lewis acid sites and makes the catalyst more active in olefin conversion.
References
F. Masters, T. Maschmeyer, Micropor. Mesopor. Mater. 142, 423 (2011)
I. Fechete, Y. Wang, J.C. Vedrine, Catal. Today 189, 2 (2012)
A. Corma, Chem. Rev. 97, 2373 (1997)
C.S. Cundy, P.A. Cox, Chem. Rev. 103, 663 (2003)
N. Kosinov, C. Liu, E.J.M. Hensen, E.A. Pidko, Chem. Mater. 30, 3177 (2018)
H. Luo, J.D. Lewis, Y.R. Leshkov, Annu. Rev. Chem. Biomol. Eng. 7, 663 (2016)
C. Gauthier, B. Chiche, A. Finiels, P. Geneste, J. Mol. Catal. 50, 219 (1989)
R. Fricke, H. Kosslick, G. Lischke, M. Richter, Chem. Rev. 210, 2303 (2000)
R. Astala, S.M. Auerbach, J. Am. Chem. Soc. 126, 1843 (2004)
M. Rutkowska, U. Diaz, A.E. Palomares, L. Chmielarz, App. Catal. B Environ. 168–169, 531 (2015)
S. Laforge, P. Ayrault, D. Martin, M. Guisnet, App. Catal. A Gen. 279, 79 (2005)
M.E. Leonowicz, J.A. Lawton, S.L. Lawton, M.K. Rubin, Science 264, 1910 (1994)
G.J. Kennedy, S.L. Lawton, M.K. Rubin, J. Am. Chem. Soc. 116, 11000 (1994)
N. Kumar, L.E. Lindfors, Appl. Catal. A 147, 175 (1996)
A. Corma, J.M. Triguero, J. Catal. 165, 102 (1997)
M.J. Verhoef, E.J. Creyghton, J.A. Peters, H.V. Bekkum, Chem. Commun. 20, 1989 (1997)
B. Tang, W. Dai, X. Sun, G. Wu, L. Li, N. Guan, M. Hunger, Chin. J. Catal. 36, 801 (2015)
W. Fan, P. Wu, S. Namba, T. Tatsumi, Angew. Chem. Int. Ed. 43, 236 (2004)
X. Ke, L. Xu, C. Zeng, L. Zhang, N. Xu, Micropor. Mesopor. Mater. 106, 68 (2007)
C.T. Brigden, D. Thompsett, C.D. Williams, Dalton Trans. 18, 2829 (2004)
Y.K. Hwang, J.S. Chang, S.E. Park, D.S. Kim, Y.U. Kwon, S.H. Jhung, J.S. Hwang, M.S. Park, Angew. Chem. Int. Ed. 44, 556 (2005)
W. Fan, R. Duan, T. Yokoi, P. Wu, Y. Kubota, T. Tatsumi, J. Am. Chem. Soc. 130, 10150 (2008)
W. Yajing, J. Wang, P. Liu, W. Zhang, J. Gu, X. Wang, J. Am. Chem. Soc. 132, 17989 (2010)
W.J. Roth, B. Gil, W. Makowski, A. Sławek, A. Korzeniowska, J. Grzybek, M. Siwek, P. Michorczyk, Catal. Sci. Technol. 6, 2742 (2016)
R.V. Jasra, J. Das, S. Unnikrishnan, A. Sakthivel, US Patent 9359216 (2016)
X. Pu, L. Shi, Catal. Today 212, 115 (2013)
C.W. Chen, W.J. Wu, X.S. Zeng, Z.H. Jiang, L. Shi, Ind. Eng. Chem. Res. 48, 10359 (2009)
W.F. Lai, R.E. Kay, M.K. Raman, US patent 7883686 (2007)
M.K. Rubin. P. Chu, U.S. Patent 4954325 (1990)
G. Liu, P. Tian, J. Li, D. Zhang, F. Zhou, Z. Liu, Micropor. Mesopor. Mater. 111, 143 (2008)
A.V. Miltenburg, J. Pawlesa, A.M. Bouzga, N. Zilkova, J. Cejka, M. Stoker, Top. Catal. 52, 1190 (2009)
Y.J. He, G.S. Nivarthy, F. Eder, K. Seshan, J.A. Lercher, Micropor. Mesopor. Mater. 25, 207 (1998)
G.T. Neumann, J.C. Hicks, ACS Catal. 2, 642 (2012)
W.J. Roth, W. Makowski, B. Marszalek, P. Michorczyk, W. Skuzaa, B. Gil, J. Mater. Chem. A 2, 15722 (2014)
X. Ouyang, Y. Wanglee, S.J. Hwang, D. Xie, T. Rea, S.I. Zones, A. Katz, Dalton Trans. 43, 10417 (2014)
J.K. Reddy, K. Motokura, T. Koyama, A. Miyaji, T. Baba, J. Catal. 289, 53 (2012)
M. Marosz, B. Samojeden, A. Kowalczyk, M. Rutkowska, M. Monika, U. Diaz, A.E. Palomares, L. Chmielarz, Materials 13, 2399 (2020)
K. Byrappa, B.V, Suresh Kumar, Asian J. Chem. 19, 4933 (2007)
J.C. Jansen, F.J.V. Gaag, H.V. Bekkum, Zeolites 4, 369 (1984)
P.A. Jacobs, H.K. Beyer, J. Valyon, Zeolites 1, 161 (1981)
Q. Dai, X. Wang, G. Chen, Y. Zheng, G. Lu, J. Catal. 207, 213 (2002)
J.A. Martens, P.A. Jacobs, Stud. Surf. Sci. Catal. 137, 633 (2001)
X. Pu, N.W. Liu, Z.H. Jiang, L. Shi, Ind. Eng. Chem. Res. 51, 13891 (2012)
P.Y. Dapsens, C. Mondelli, J.P. Ramirez, Chem. Soc. Rev. 44, 7025 (2015)
G. Li, E.A. Pidko, ChemCatChem 11, 134 (2019)
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Reddy, J.K., Mantri, K., Lad, S. et al. Synthesis of Ce-MCM-22 and its enhanced catalytic performance for the removal of olefins from aromatic stream. J Porous Mater 27, 1649–1658 (2020). https://doi.org/10.1007/s10934-020-00940-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10934-020-00940-x