
Abstract
Herein, sodium alginate (SA) and hydrolyzed collagen (HC) blend films incorporating propolis extract (PE) (1–4 wt%) were prepared. A thorough morphological, structural and property-related evaluation has been provided. Micro-structurally, the incorporation of low PE concentrations resulted in more uniform SA/HC films. The FTIR results confirmed no modifications in the film’s functional groups, and the crystallinity degrees (XRD) of the films ranged from 4.1 to 5.6%. The incorporation of PE caused an increase in tensile strength, from 7.9 MPa (SA/HC) to 16.3 MPa (SA/HC/PE3%). Additionally, PE incorporation reduced the water affinity of the films, leading to a reduction in their moisture content. A type III moisture sorption isotherm was observed for all films at 25 °C, and the Smith’s model provided the best fit, suggesting that SA, HC and PE presented good miscibility, reducing the availability of functional groups for adsorption of water molecules. More importantly, the incorporation of PE improved the antioxidant activity of the films from 3.43% (SA/HC) to 22.5% (SA/HC/PE4%), and provided an exceptional UV light blocking ability. Overall, green PE can be considered an interesting option for the enhancement of SA/HC bioactive film performance.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Food packaging produced using petroleum-based polymers has generated enormous concern as being a potential environmental pollutant. In this sense, there is a tendency to replace these polymers with renewable and/or biodegradable packaging materials, such as those based on polysaccharides and proteins [1,2,3]. Numerous biologically-based polymers have shown promise for the production of films and coatings with food packaging applications, such as starch, chitosan, alginate, pectin, gelatin and collagen [4,5,6,7,8,9].
Sodium alginate (SA) is a natural polysaccharide extracted from seaweed and is composed of linear polyuronic acid (1 → 4) linked by β-d-mannuronic and α-l-guluronate, which has good biodegradability, high compatibility and the ability to form films [10,11,12]. However, pure SA-based films still have relatively weak mechanical and water vapor barrier properties, limiting their application in food packaging [12, 13]. SA/gelatin or SA/hydrolyzed collagen blends, which are typical polysaccharide/protein systems, have demonstrated good ability to form films with superior mechanical properties [13]. Hydrolyzed collagen (HC) is a mixture of amino acids joined to form elongated fibrils. They are obtained from animal bones and skins or even from industrial leather waste, considered as their main structural protein [14, 15]. HC is a biocompatible and biodegradable material with potential for the preparation of films [14, 16, 17].
The incorporation of active compounds with antioxidant and/or antimicrobial activity is a promising alternative for the development of active films and coatings in food packaging [18]. Propolis is a resinous mixture of substances made by bees, mainly composed of vegetable resins and balms, waxes, essential and aromatic oils, pollen and other substances [18,19,20]. Green propolis is produced by Apis mellifera bees, which collect nectar from the Baccharis dracunculifolia plant, which is very popular in Brazil [21, 22]. The green propolis extract (PE) has a phenolic composition that includes p-coumaric acid and artepilin C, contributing to its effective antioxidant capacity [23]. Due to these characteristics, PE can be considered an outstanding bioactive ingredient in biopolymer matrices for the production of antioxidant films [24].
Packaging films with antioxidant compounds can interact with foods and/or their environment, either by capturing pro-oxidant compounds or by releasing antioxidant molecules in packaged foods [1, 24, 25]. These active films can contribute to increasing the shelf-life of foods, as degradation is driven by oxidation reactions [26]. The antioxidant potential of films containing PE is still little explored [24]. Some examples of bio-based matrices incorporating PE include carrageenan [27], starch [27,28,29], chitosan [30], pectin [31], rice starch/carboxymethyl chitosan [32] and poly(lactic) acid [33]. In the majority of works found in literature on the subject, different alcoholic/hydroalcoholic PEs have been used, which ultimately contain compounds of low water solubility and/or are incompatible with biopolymer matrices. The result may be development of films with weakened mechanical properties from the incorporation of higher PE loads.
Faced with the enormous scarcity of the use of aqueous green PE as a bioactive ingredient for bio-based food packaging, this study aimed at the development of films based on SA and HC blends as well as incorporating different concentrations of PE (1, 2, 3 and 4 wt%). Unlike what has traditionally been explored in the literature, the fundamental hypothesis of this study was that the use of the green PE aqueous fraction would enable the development of films with greater compatibility between the components, allowing the improvement of the materials’ properties. Therefore, the effects of PE on the structural, physicochemical and antioxidant properties of the SA/HC films were assessed for the first time. We believe that this study could be a bottom line for the development of sustainable antioxidant films with better general performance from the incorporation of aqueous green PE.
Material and Methods
Material
Sodium alginate (SA) with a viscosity (2% solution at 25 °C) of 2 cps, and glycerol was purchased from Dinâmica Química Contemporânea Ltda, Indaiatuba/SP, Brazil; hydrolyzed collagen (HC) was procured from NaturalLife, São José do Rio Preto/SP, Brazil; and dried hydrosoluble green propolis extract (PE) (80% purity) was donated by Apis Flora Industrial e Comercial Ltda, Ribeirão Preto/SP, Brazil.
Preparation of Film-Forming Solution and Film Samples
Active films from sodium alginate/hydrolyzed collagen with propolis extract (SA/HC/PE) were prepared employing the casting method. SA film-forming solutions (4% w/w) were prepared via dissolving in distilled water previously containing glycerol (30 g/100 g of SA) and hydrolyzed collagen (10 g/100 g of SA). This proportion of HC was defined from a preliminary study [34]. Different solutions were prepared for the evaluation of the propolis extract effect (0, 1, 2, 3 and 4% w/w, considering the total SA and HC mass as the calculation basis). The film-forming solutions were previously homogenized at 4000 rpm for 5 min in an ultra-turrax (IKA T18 basic). Then, they were heated to 80 °C for 20 min, followed by another homogenization at 4000 rpm for 5 min in the ultra-turrax, and treated in an ultrasound bath for 15 min. After this step, 50 g of the film-forming solution was poured into polystyrene Petri dishes (14 cm diameter). The solutions were dried in a forced-air oven (Ethik Technology, Vargem Grande Paulista/SP, Brazil) at 40 °C for 24 h. The films were identified as control samples: SA/HC, SA/HC/PE1%, SA/HC/PE2%, SA/HC/PE3% and SA/HC/PE4%. All of the films were placed in an air-conditioned laboratory at 25 °C and 75% RH before the characterization test.
Film Characterization
Surface Morphology
Analysis of the surface and cross-section (fractured with liquid nitrogen) of the obtained films was performed via scanning electron microscopy (SEM) (Leo 440i, LEO Electron Microscopy/Oxford, Cambridge, England) at an acceleration voltage of 15 kV, 50 pA current and magnitude of 1000 × (surface) and 2500 × (cross-section). The samples were fixed in metallic support with the aid of double-sided carbon tape and covered with a thin layer of gold in a sputter coater (SC7620, VG Microtech, Kent, United Kingdom).
Fourier-Transform Infrared Spectroscopy (FTIR)
FTIR spectra of the films were recorded by the attenuated total reflection method (ATR) using a Thermo Scientific spectrophotometer (Nicolet Continuum, Madison, USA). The spectra were recorded between a range of 4000–650 cm−1 with a resolution of 4 cm−1 and 128 scans.
Thermogravimetric Analysis (TGA)
Thermal stability of the films was determined in a Thermogravimetric Analyzer (Mettler Toledo, TGA/DSC1, Schwerzenbach, Switzerland). For this, 10 mg of each film sample was heated from 25 to 600 °C in a nitrogen atmosphere at a flow rate of 50 mL min−1. All experiments were conducted at 20 °C min−1 and the TG/DTG curves were also provided.
X-ray Diffraction (XRD)
X-ray diffraction analysis of the films was performed by an X-ray analyzer (X'Pert-MPD, Philips, Almelo, Netherlands), using Cu Kα radiation (1.54056 Å) at a scan rate of 0.033333°/s (step = 0.04° and time per step = 1.2 s), with the accelerated voltage of 40 kV and the applied current of 40 mA, ranging from 5 to 60°. The degree of crystallinity was estimated from the deconvolution of the peaks and calculation of the ratios between the areas of the crystalline regions and the total area (crystalline and amorphous). This was carried out using the Fityk software.
Thickness
Average film thickness was measured using a digital micrometer (Mitutoyo Co., Kawasaki-Shi, Japan), with a resolution of 0.1 µm. Measurements were performed at 5 random locations from 5 specimens and from the same film formulation [35].
Mechanical Properties
Tensile strength (MPa), elongation at break (%) and modulus of elasticity (MPa) of the films were determined using a universal testing machine (Instron, 5966-E2, Norwood, USA). For this, the samples were cut to a width of 15 mm in high precision equipment (RDS-100-C, ChemInstruments, OH, USA). Then, they were conditioned for 48 h at 23 ± 2 °C and 50 ± 5% RH. The tests were performed with a 1 kN load cell, at a speed of 12 mm min−1 and with initial distances between grips equal to 50 mm. For each sample, 5 repetitions were performed [36].
Moisture Sorption Isotherms
A Dynamic Vapor Sorption system (DVS-002, Surface Measurements System, London, UK), with controlled atmosphere, was employed for determination of moisture sorption isotherms. Each film sample (0.5 cm × 0.5 cm) was previously placed in a vacuum oven (45 °C) for 18 h. Then, the samples were cooled down to 25 °C in a desiccator (~ 20 min), and 50 mg of each sample was analyzed. During the analysis, the following RH profile was applied: 0.00, 10.56, 21.11, 31.67, 42.22, 52.78, 63.33, 73.89, 84.44, and 95% RH. The RH step lasted until reaching mass equilibrium, and the moisture sorption was calculated as the gained water mass (grams) per grams of dry film at the adopted RH.
To understand the moisture sorption behaviour, experimental data were adjusted to different models (Table 1). The determination of constant parameters was carried out using Excel®. The generalized reduced gradient (GRG) method from the Excel Solver was carried out to estimate the GAB and BET model parameters by minimizing the Residual Standard Deviation (Sres) (Eq. 1). The other models were simply linearized, allowing parameter determinations from angular and linear coefficients. To attest for the fitting performance, determination coefficients (R2) and Residual Standard Deviation (Sres) were provided. Models in which R2 > 0.98 and Sres < 0.2 were considered as good fit. This was calculated according to the following formula:
where: Sres is the Residual Standard Deviation, Yexp denotes the experimental value, Ysim demonstrates the predicted value (model considered) and “n” shows the amount of experimental data.
Moisture Content Immediately After Film Preparation
After preparing the film samples, moisture contents (MC) were evaluated using Eq. (2). The initial masses (wi) of the samples were determined, and then they were subjected to 105 °C for 24 h in an oven (Ethik Technology, Vargem Grande Paulista, Brazil). After cooling the samples, the final masses (wf) were measured. The MC values (%) were determined using 4 repetitions. Below, is the formula used for calculations:
Water Vapor Permeability (WVP)
Water vapor transmission rate (WVTR), expressed in g m−2 day−1, was determined using capsules with 50 cm2 permeation area. Tests were carried out at 25 °C and 75% RH in an air-conditioning chamber (Weiss Technik, Reiskirchen, Germany), with anhydrous calcium chloride desiccant inside the capsules. The change of mass was monitored with an analytical balance (Mettler Toledo, Columbus, Ohio, USA) of 10–4 g resolution. WVTR was estimated from the slope of the “mass vs. time” curve [37], in 4 repetitions. Finally, WVP (g m−1 s−1 Pa−1) of the film was calculated according to Eq. (3):
where: e is the specimen average thickness (µm), ps shows the water vapor saturation pressure (3168 Pa at 25 °C), RH1 demonstrates the relative humidity of the chamber (75% = factor 0.75). RH inside the capsule was considered to be 0 (zero).
Light Transmission
Light transmission through the films was assessed using a double-beam UV–visible spectrophotometer (Analytik Jena—Specord 210), with a wavelength ranging from 200 to 800 nm and a scanning speed of 120 nm min−1 [38]. The tests were performed in triplicate.
Antioxidant Activity
The antioxidant activity of the films was evaluated using DPPH (2,2-diphenyl-1-picrylhydrazyl) free radical scavenging assay [30]. Briefly, 25 mg of film sample was dissolved in 3 mL of distilled water and then mixed with 1 mL methanolic DPPH solution at a concentration of 1 mM. The mixture was vortexed and incubated in the dark at 25 °C for 30 min. The absorbance was measured at 517 nm. The percentage of DPPH free radical scavenging activity was determined using Eq. (4):
where: AbsDPPH is the absorbance value at 517 nm of the DPPH methanolic solution and Absextract demonstrates the absorbance value at 517 nm for the sample extracts. The tests were performed in triplicate.
Statistical Analysis
The results were statistically evaluated by means of analysis of variance (ANOVA) and Tukey’s test to compare averages. Differences were considered to be statistically significant if p < 0.05.
Results and Discussion
Surface Morphology
The microstructure confirmed by SEM photos of all the films is shown in Fig. 1. The SA/HC films demonstrated good homogeneity both on the surface and on the cross-section. No streaks or micropores were observed, which proves very good compatibility between the film components. The presence of inter- and intra-molecular hydrogen bonds between biopolymers promotes the formation of a homogeneous structure concerning the film-forming matrix [39]. As the concentration of the green propolis additive increased, the structure became spongier. The highest concentration of propolis resulted in microcracks in the cross-section, while the surface became granular. The addition of phenolic compounds and natural pigments to the film may promote the formation of cracks [40].

Scanning electron micrography of sodium alginate/hydrolyzed collagen with propolis extract. Evaluation of the composite film morphology, in which a, c, e, g and i refer to surface images with × 1000 magnification; and b, d, f, h and j refer to the cross-sectional images, with × 2500 magnification. a and b: SA/HC, c and d: SA/HC/PE1%, e and f: SA/HC/PE2%, g and h: SA/HC/PE3%, and i and j: SA/HC/PE4%
FTIR Spectroscopy
FTIR spectra of the pure PE and SA/HC/PE-based films are presented in Fig. 2. For the PE spectra, it is possible to note a typical band around 3360 cm−1 related to the –OH groups. The peaks at 1635 cm−1 and 1556 cm−1 may be due to the stretching vibration of C=C from aromatic ring deformations of phenolic compounds [41], or due to the C=O stretching vibration of caffeic acid and its derivatives [42]. The peak at 1250 cm−1 may be related to the C–H wagging vibration of phenolic compounds or to C–O groups of polyols [43], and around 980 cm−1, related to wagging –CH2 of monoterpenes [44].

a FTIR spectra and (of sodium alginate/hydrolyzed collagen with propolis extract
Considering SA/HC and SA/HC/PE of the film spectra, an intense band around 3344 cm−1 is also present, related to stretching vibration of –OH bonds. The peaks at approximately 1600 and 1408 cm−1 are typical of asymmetric and symmetric stretching vibrations of C–O in the COO− group [34, 45, 46]. Additionally, the weak peak at approximately 1080 cm−1 is related to C–O and C–C stretching vibrations of pyranose rings [47], while the sharp peak observed at 1028 cm−1 can be attributed to the antisymmetric stretch of C–O–C [48]. Another weak peak at 818 cm−1 can be ascribed to mannuronic acid residues [47, 49].
Overall, it has been found that the FTIR spectra of all films containing PE had a profile very similar to the pure SA/HC films, that is, bands typical of pure PE were not observed due to the low concentration and superimposing signals on similar wavenumbers. Furthermore, the presence of new bands was unnoticed, suggesting that no new chemical bonds were formed and that the SA/HC/PE interactions were only physical. This is desirable behaviour, as it would allow the release of PE bioactive compounds from the films to the food potentially packaged with this material.
Thermogravimetric Analysis (TGA)
In Fig. 3, the thermogravimetric characterization is shown of SA/HC and SA/HC/PE films. The curves depict the mass loss (Fig. 3a) of the blend samples during the thermal screening. The derivative of the thermogravimetric curves can also be observed (Fig. 3b). Degradation of all samples studied occurred in 2 thermal events, which is typical for blend films prepared from sodium alginate [50].

TGA a and DTGA b curves of sodium alginate/hydrolyzed collagen with propolis extract.
The first thermal event present in all films was detected at a temperature inferior to 100 °C. Such an occurrence is related to free water evaporation from the films, which was around 20% at 75% RH, storage condition (Fig. 5a). The second thermal event is associated with SA and HC thermal disintegration, which occurs in a similar range, justifying the presence of a single thermal event for the blend [51, 52]. The increment of PE incorporation on the sodium HC/SA matrix, within the range of 0–4%, decreased the average mass loss rate (Fig. 3a) of the films up to a temperature of approximately 230 °C. A similar effect was reported in the literature regarding the evaluation of SiO2 incorporation into SA/HC blends [52].

XRD pattern of sodium alginate/hydrolyzed collagen with propolis extract
In Table 2, data are presented from the 2 thermal events registered on the thermograms of Fig. 3. The initial degradation (Tonset) and maximum decomposition (Tmax) temperatures of the control SA/HC film are similar to those reported in the literature [52, 53]. As for other formulations, there were no important changes in the thermal behaviour of SA/HC/PE films, regardless of PE concentration. However, a trend showing a slight decrease in the Tmax was observed as a result of PE inclusion in the polymeric matrix (Table 2). This might be due to a disruption in the polymer matrix structure caused by the presence of propolis chains.
X-ray Diffraction (XRD)
In Fig. 4, the XRD patterns are illustrated of all samples from the present study. The general aspect of the diffractograms reveals the semi-crystalline nature of the prepared materials. All formulated blends, including the SA/HC control, present a similar XRD pattern with a sharp crystalline peak at 2θ ≈ 14° and a broad one at around 2θ ≈ 22°, which are characteristic of SA/HC blends [34, 52]. Through the deconvolution of the peaks, it was possible to estimate the degree of crystallinity for each sample from the calculation of the crystalline and amorphous areas (Table 3).
All the film samples presented very low degrees of crystallinity, highlighting its mostly amorphous nature. Different degrees of crystallinity have been reported for SA, from values as low as 0.06% [54], to relatively high values around 35% [55]. This variability is common, especially in the case of natural polymers, the structural characteristics of which are dependent on factors such as sources, extraction and preparation methods [11].
Overall, the diffractogram patterns have similar aspects and degrees of crystallinity, with a slightly higher value for the SA/HC control sample. Hence, it is not possible to claim that the incorporation of PE altered the crystallinity of the films prepared in this study. Differently, Suriyatem et al. found that the incorporation of PE in rice starch/carboxymethyl chitosan films reduced the crystallinity of the blends in the case of a 2.5% concentration of the additive [32].
Thickness and Mechanical Properties
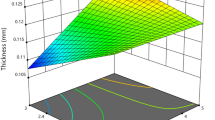
The thickness and mechanical properties of the SA/HC-based films are shown in Table 4. The addition of PE to the SA/HC film caused a tendency to increase the thickness, but only the incorporation of 3 and 4% of PE provided a statistically significant increase in thickness. Moreover, the SA/HC films incorporated with PE displayed different mechanical properties than the control samples. It was observed that the incorporation of PE presented a very significant influence on the tensile strength (TS) of the films. For example, comparing the SA/HC and SA/HC/PE3% samples, there was an increase by approximately 106% in TS. Additionally, the modulus of elasticity (ME) of the films also showed an increase due to the incorporation of PE, indicating that the stiffness of the films also increased.
The reported phenomenon can be attributed to the intermolecular interactions, such as hydrogen bonding, between the components presenting polar groups of PE (e.g., balsams containing flavonoids, phenolic acids and their extracts), and hydrophilic groups of SA and HC. As a result, the formation of stronger interfacial adhesion between the biopolymers and PE components [30, 56], was achieved. It is noteworthy that water-soluble PE was used in this present study. That is, its components present a mostly polar nature, providing an excellent compatibilization in the polymer matrix, resulting in higher TS as well as ME. Upon the incorporation of PE, an apparent decrease in the elongation at break (EB) was noted (Table 4). However, no statistically significant differences (p > 0.05) were observed. This is an important result, as it suggests that the incorporation of PE can improve the TS of the films without significant losses of EB. A similar result was reported for the incorporation of propolis into chitosan [30], and hydroxypropyl methylcellulose-based films [56].
Moisture Content and Water Vapor Permeability
The incorporation of PE significantly (p < 0.05) decreased the moisture content of the films compared to the control (SA/HC) (Table 5). Similar results were found for chitosan/zein/essential oil films functionalized with propolis polyphenols [57]. The decrease in moisture content of the films is mainly attributed to the interactions between biopolymers and PE, which reduces the accessibility to free hydroxyl groups, affecting its water absorption capacity [58, 59]. It is noteworthy that the moisture contents given in Table 5 was determined after film preparation and may be related to the solvent evaporation time and the ambient relative humidity at the moment of analysis. Moisture sorption studies were also performed and discussed subsequently.
The WVP of the control film (SA/HC) was 2.65 ± 0.05 × 10–10 g m–1 s–1 Pa–1 (Table 4). The addition of PE significantly increased (p < 0.05) the WVP of the films, reaching a maximum value of 4.48 ± 0.21 × 10–10 g m–1 s–1 Pa–1 for the SA/HC/PE3% sample. Even having acted as a kind of mechanical reinforcement to the films, the incorporation of bioactive compounds present in the PE may have contributed to the increase in free volume regarding the polymer matrix, causing increased WVP. The incorporation of small molecules, being located between macromolecules, can inevitably generate voids around them [31]. Moreover, the addition of higher PE loads resulted in small microstructural discontinuities, possibly to a partial agglomeration of undissolved material, as seen in the cross-section SEM images (films with 3% PE), generating preferential diffusion paths and, therefore, favoring the higher WVP value for this sample.
The aforementioned results are in accordance with those reported in the literature, in which WVP was affected due to the presence of cracks and irregularities in the polymer matrix, resulting from the addition of propolis [60]. The increase in WVP of biopolymer-based films due to the addition of PE was reported for poly(lactic acid) [61], and rice starch/carboxymethyl chitosan [32] biopolymer-based films. That is, following a trend very similar to that reported in this study. However, SA-based films reinforced with nano-SiO2 incorporating aqueous PE showed no statistically significant loss of water vapor barrier [62]. Despite the changes that occurred in WVP from the incorporation of PE in this study, it appears that its value can still be considered high for applications such as packaging of moisture-sensitive foods.
Moisture Sorption Isotherms
For a more robust analysis of the interactions between ambient humidity and the films prepared in this study, moisture sorption isotherms were obtained in high-precision equipment and reported in Fig. 5. Water activities (aw) were evaluated from 0 to 0.95, that is, the films were submitted to extreme relative humidity conditions. As responses, the absolute moisture content of the films was recorded. In Fig. 5(a), the profiles obtained from each sample are compared. It can be seen that for values of aw < 0.75, the control film (SA/HC) showed a tendency towards retaining a higher amount of moisture. For values of aw > 0.8, the samples with higher concentrations of PE demonstrated the retention of higher absolute humidity. Therefore, the moisture content values reported in Table 4 could have been different depending on the relative humidity and temperature conditions of the measurement environment.
Overall, all isotherms presented a similar pattern. Moisture content (MC) remained at very low values and increased very little until values reached aw < 0.4, with virtually equivalent values for all samples. An increase in MC only became noticeable for aw values ranging from 0.4 to 0.95, as similarly reported for SA/HC/SiO2 composites [5]. This behavior refers to type III isotherms [63], suggesting that the attractive forces between adsorbed water and functional groups from film matrix are lower than the attractive forces between the water molecules in the liquid state. This result allows to highlight that SA, HC and PE formed a film matrix with good compatibility, i.e., with reduced availability of functional groups for adsorption of water molecules [5].
Classic models of moisture sorption isotherms were fitted to the experimental results of this study. In Fig. 5 (b–f), a comparison is provided between each experimental point and Smith’s model, which was considered the best fit (R2 > 0.98 and Sres < 0.2) for the entire range of aw. This model was proposed to reproduce the final curved portion of the water sorption isotherm of high molecular weight polymers [64]. In Table 6, the determined parameters are presented for all models, with R2 and Sres. Traditional models, such as the GAB and BET, did not satisfactorily fit the data to the detriment concerning some particularities of each case. For example, a type III isotherm does not have a “knee point” as in the sigmoidal profiles, i.e., there is no restricted multilayer formation [65]. Furthermore, in this study, a very wide aw range was considered and the majority of models reproduced the data satisfactorily in limited ranges. This may explain the not-so-adequate fit of the GAB and BET models to the experimental data.
Light Transmission
The UV–visible light transmission spectra of the SA/HC/PE-based films are shown in Fig. 6. The control film (SA/HC) presented a high barrier to UV light up to approximately 350 nm, with values close to 0. This behaviour can be attributed to the aromatic amino acids present in collagen, which are chromophores that can absorb low wavelength UV light [66,67,68,69]. Moreover, the n → π ∗ transitions in carbonyl groups of SA, with a maximum absorption around 283 nm, and the high absorption coefficient of C=O bond, below 230 nm, due to π → π ∗ transition [31, 70], may also have contributed to this effective UV light barrier of the films at low wavelengths. Light transmission gradually increased as the wavelength was increasing to the visible range. Interestingly, the incorporation of PE extended the SA/HC light barrier near 0 to approximately 400 nm. Furthermore, a gain in visible light barrier was observed, in which the barrier increases with increasing PE concentration. This behaviour is attributed to the light absorption capacity of the phenolic compounds present in PE [59], which present numerous chromophores groups, such as C=C and C=O, displaying a strong light absorption capacity [24, 31].

Moisture sorption isotherm profiles of sodium alginate and collagen hydrolyzed with propolis extract films
Antioxidant Activity
The control film (SA/HC) presented a DPPH radical scavenging value of 3.43 ± 0.87% (Fig. 7). Although this film does not contain an active extract, its antioxidant activity may be due to a small proportion of polyphenolic compounds present in alginates from brown algae [71] and biologically active peptides from hydrolyzed collagen [72]. On the other hand, the PE has a powerful antioxidant activity, due to the presence of phenolic compounds. Thus, the addition of this extract can significantly increase the antioxidant activity of biopolymer-based films [24].

Light transmission (%) of sodium alginate/hydrolyzed collagen with propolis extract

DPPH free radical scavenging activity of sodium alginate/hydrolyzed collagen with propolis extract
Incorporation of the propolis extract to the SA/HC films resulted in a significant increase (p < 0.05) in the antioxidant capacity of the films. The SA/HC films exhibited maximum DPPH radical scavenging values of 22.5% (SA/HC/PE4%). This result is attributed to the higher concentration of phenolic compounds from the propolis extract present in the film. The main active compounds of green propolis are quercetin, artepelin C, p-coumaric acid and cinnamic acid [73, 74]. Corroborating these results, a proportional increase in the antioxidant capacity as a function of PE concentration was also observed for pectin [31], starch [29], rice starch/carboxymethyl chitosan [32], carrageenan/starch [27], chitosan [30] and poly(lactic acid) [33] films. Due to the great variability in the composition of the different types of available PE, there is still much to be explored in this field. For example, the antioxidant activity of films containing green PE are still considered scarce, as discussed in a recent review article [24]. Still, these materials have great potential for applications as active food packaging, resulting in higher quality and increased shelf-life, especially for foods that can undergo oxidative reactions.
Conclusions
This study aimed to develop active films based on sodium alginate (SA) and hydrolyzed collagen (HC) incorporating different proportions (1–4% w/w) of an aqueous propolis extract (PE). FTIR spectra of all films containing PE had a similar profile to the pure SA/HC films. The presence of new bands was unnoticed, suggesting that no new chemical bonds were formed and that the SA/HC/PE interactions were only physical. There was an increase by approximately 106% in tensile strength (comparing SA/HC and SA/HC/PE3% samples); and the modulus of elasticity also showed an increase from the incorporation of PE, while elongation at break decreased. Furthermore, the incorporation of PE extended the UV/Vis light barrier effect of the films, and resulted in a significant increase of antioxidant activity. In conclusion, since the mechanical properties can be improved; and also, light barrier and antioxidant properties are imparted to SA/HC/PE3% film, this formulation can be considered the most attractive option for further studies in contact with foods. The film developed could provide higher quality and increased shelf-life for packaged foods that can undergo, for example, oxidative reactions. Subsequent studies on the contact of these films with foods will be fundamental to consolidating such applications.
References
Li T, Xia N, Xu L et al (2021) Preparation, characterization and application of SPI-based blend film with antioxidant activity. Food Packag Shelf Life 27:100614. https://doi.org/10.1016/j.fpsl.2020.100614
Li Y, Chen K, Yang Q et al (2021) Development and characterization of novel antioxidant films based on chitosan and Maillard reaction products. LWT-Food Sci Technol 141:110886. https://doi.org/10.1016/j.lwt.2021.110886
Vianna TC, Marinho CO, Júnior LM et al (2021) Essential oils as additives in active starch-based food packaging films: a review. Int J Biol Macromol. https://doi.org/10.1016/j.ijbiomac.2021.05.170
Marangoni Júnior L, Vieira RP, Jamróz E, Anjos CAR (2021) Furcellaran: an innovative biopolymer in the production of films and coatings. Carbohydr Polym 252:117221. https://doi.org/10.1016/j.carbpol.2020.117221
Marangoni Júnior L, da Silva RG, Vieira RP, Alves RMV (2021) Water vapor sorption and permeability of sustainable alginate/collagen/SiO2 composite films. LWT 152:112261. https://doi.org/10.1016/j.lwt.2021.112261
Michelin M, Marques AM, Pastrana LM et al (2020) Carboxymethyl cellulose-based films: effect of organosolv lignin incorporation on physicochemical and antioxidant properties. J Food Eng 285:110107. https://doi.org/10.1016/j.jfoodeng.2020.110107
Mohamed SAA, El-Sakhawy M, El-Sakhawy MA-M (2020) Polysaccharides, protein and lipid -based natural edible films in food packaging : a review. Carbohydr Polym 238:116178. https://doi.org/10.1016/j.carbpol.2020.116178
Marangoni Júnior L, Fozzatti CR, Jamróz E et al (2022) Biopolymer-based films from sodium alginate and citrus pectin reinforced with SiO2. Mater 15:3881
Marinho CO, Vianna TC, Cecci RRR et al (2022) Effect of water kefir grain biomass on chitosan film properties. Mater Today Commun 32:103902. https://doi.org/10.1016/j.mtcomm.2022.103902
Senturk Parreidt T, Müller K, Schmid M (2018) Alginate-based edible films and coatings for food packaging applications. Foods 7:170
Marangoni Júnior L, da Silva RG, Anjos CAR et al (2021) Effect of low concentrations of SiO2 nanoparticles on the physical and chemical properties of sodium alginate-based films. Carbohydr Polym 269:118286. https://doi.org/10.1016/j.carbpol.2021.118286
Chen J, Wu A, Yang M et al (2021) Characterization of sodium alginate-based films incorporated with thymol for fresh-cut apple packaging. Food Control 126:108063. https://doi.org/10.1016/j.foodcont.2021.108063
Yang L, Yang J, Qin X et al (2020) Ternary composite films with simultaneously enhanced strength and ductility: effects of sodium alginate-gelatin weight ratio and graphene oxide content. Int J Biol Macromol 156:494–503. https://doi.org/10.1016/j.ijbiomac.2020.04.057
Pinto TDS, Rodrigues PN, Marinho LE et al (2019) Self-assembled hybrid nanocomposite films of carbon dots and hydrolyzed collagen. Mater Chem Phys 230:44–53. https://doi.org/10.1016/j.matchemphys.2019.03.060
Chuysinuan P, Thanyacharoen T, Thongchai K et al (2020) Preparation of chitosan/hydrolyzed collagen/hyaluronic acid based hydrogel composite with caffeic acid addition. Int J Biol Macromol 162:1937–1943. https://doi.org/10.1016/j.ijbiomac.2020.08.139
Walczak M, Michalska-Sionkowska M, Kaczmarek B, Sionkowska A (2020) Surface and antibacterial properties of thin films based on collagen and thymol. Mater Today Commun 22:100949. https://doi.org/10.1016/j.mtcomm.2020.100949
Valencia GA, Luciano CG, Lourenço RV et al (2019) Morphological and physical properties of nano-biocomposite films based on collagen loaded with laponite®. Food Packag Shelf Life 19:24–30. https://doi.org/10.1016/j.fpsl.2018.11.013
Reyes LM, Landgraf M, Sobral PJA (2021) Gelatin-based films activated with red propolis ethanolic extract and essential oils. Food Packag Shelf Life 27:100607. https://doi.org/10.1016/j.fpsl.2020.100607
Correa-Pacheco ZN, Bautista-Baños S, de LorenaRamos-García M et al (2019) Physicochemical characterization and antimicrobial activity of edible propolis-chitosan nanoparticle films. Prog Org Coatings 137:105326. https://doi.org/10.1016/j.porgcoat.2019.105326
Moreno MA, Vallejo AM, Ballester A-R et al (2020) Antifungal edible coatings containing Argentinian propolis extract and their application in raspberries. Food Hydrocoll 107:105973. https://doi.org/10.1016/j.foodhyd.2020.105973
Machado BA, Silva RP, Barreto GD et al (2016) Chemical composition and biological activity of extracts obtained by supercritical extraction and ethanolic extraction of brown, green and red propolis derived from different geographic regions in Brazil. PLoS ONE 11:e0145954
Cruz AIC, da Costa M, Mafra JF et al (2021) A sodium alginate bilayer coating incorporated with green propolis extract as a powerful tool to extend Colossoma macropomum fillet shelf-life. Food Chem 355:129610. https://doi.org/10.1016/j.foodchem.2021.129610
Cavalaro RI, da Cruz RG, Dupont S et al (2019) In vitro and in vivo antioxidant properties of bioactive compounds from green propolis obtained by ultrasound-assisted extraction. Food Chem X 4:100054. https://doi.org/10.1016/j.fochx.2019.100054
Yong H, Liu J (2021) Active packaging films and edible coatings based on polyphenol-rich propolis extract: a review. Compr Rev Food Sci Food Saf 20:2106–2145. https://doi.org/10.1111/1541-4337.12697
Otoni CG, Avena-Bustillos RJ, Azeredo HMC et al (2017) Recent advances on edible films based on fruits and vegetables—a review. Compr Rev Food Sci Food Saf 16:1151–1169. https://doi.org/10.1111/1541-4337.12281
de Lima Silva ID, de Moraes LE, Caetano VF et al (2021) Development of antioxidant active PVA films with plant extract of Caesalpinia ferrea Martius. LWT 144:111215. https://doi.org/10.1016/j.lwt.2021.111215
Chang-Bravo L, López-Córdoba A, Martino M (2014) Biopolymeric matrices made of carrageenan and corn starch for the antioxidant extracts delivery of Cuban red propolis and yerba mate. React Funct Polym 85:11–19. https://doi.org/10.1016/j.reactfunctpolym.2014.09.025
Costa SS, Druzian JI, Machado BAS et al (2014) Bi-Functional biobased packing of the cassava starch, glycerol, Licuri Nanocellulose and red propolis. PLoS ONE 9:e112554
de Araújo GKP, de Souza SJ, da Silva MV et al (2015) Physical, antimicrobial and antioxidant properties of starch-based film containing ethanolic propolis extract. Int J Food Sci Technol 50:2080–2087. https://doi.org/10.1111/ijfs.12869
Siripatrawan U, Vitchayakitti W (2016) Improving functional properties of chitosan films as active food packaging by incorporating with propolis. Food Hydrocoll 61:695–702. https://doi.org/10.1016/j.foodhyd.2016.06.001
Júnior LM, de Ávila Gonçalves S, da Silva RG et al (2022) Effect of green propolis extract on functional properties of active pectin-based films. Food Hydrocoll 131:107746. https://doi.org/10.1016/j.foodhyd.2022.107746
Suriyatem R, Auras RA, Rachtanapun C, Rachtanapun P (2018) Biodegradable rice starch/carboxymethyl chitosan films with added propolis extract for potential use as active food packaging. Polym 10:954
Ulloa PA, Vidal J, Lopéz de Dicastillo C et al (2019) Development of poly(lactic acid) films with propolis as a source of active compounds: biodegradability, physical, and functional properties. J Appl Polym Sci 136:47090. https://doi.org/10.1002/app.47090
Marangoni Júnior L, Rodrigues PR, da Silva RG et al (2021) Sustainable packaging films composed of sodium alginate and hydrolyzed collagen: preparation and characterization. Food Bioprocess Technol 14:2336–2346. https://doi.org/10.1007/s11947-021-02727-7
ISO-4593 (1993) Plastics: film and sheeting determination of thickness by mechanical scanning. Switzerland. 2
ASTM-D882 (2018) Standard Test Method for Tensile Properties of Thin Plastic Sheeting. West Conshohocken. 12
ASTM-E96/E96M (2016) Standard Test Methods for Water Vapor Transmission of materials, West Conshohocken. 14
ASTM-E-1348 (2015) Standard test method for transmittance and color by spectrophotometry using hemispherical geometry. West Conshohocken. 3
Vargas-Torrico MF, von Borries-Medrano E, Aguilar-Méndez MA (2022) Development of gelatin/carboxymethylcellulose active films containing Hass avocado peel extract and their application as a packaging for the preservation of berries. Int J Biol Macromol 206:1012–1025. https://doi.org/10.1016/j.ijbiomac.2022.03.101
Sirisha Nallan Chakravartula S, Lourenço RV, Balestra F et al (2020) Influence of pitanga (Eugenia uniflora L.) leaf extract and/or natamycin on properties of cassava starch/chitosan active films. Food Packag Shelf Life 24:100498. https://doi.org/10.1016/j.fpsl.2020.100498
Barud HDS, de Araújo Júnior AM, Saska S et al (2013) Antimicrobial brazilian propolis (epp-af) containing biocellulose membranes as promising biomaterial for skin wound healing. Evidence-Based Complement Altern Med 2013:703024. https://doi.org/10.1155/2013/703024
Sharaf S, Higazy A, Hebeish A (2013) Propolis induced antibacterial activity and other technical properties of cotton textiles. Int J Biol Macromol 59:408–416. https://doi.org/10.1016/j.ijbiomac.2013.04.030
Silva AJ, Silva JR, de Souza NC, Souto PCS (2014) Membranes from latex with propolis for biomedical applications. Mater Lett 116:235–238. https://doi.org/10.1016/j.matlet.2013.11.045
Oliveira RN, Moreira APD, da Thiré RM, SM, et al (2017) Absorbent polyvinyl alcohol–sodium carboxymethyl cellulose hydrogels for propolis delivery in wound healing applications. Polym Eng Sci 57:1224–1233. https://doi.org/10.1002/pen.24500
Voo W-P, Lee B-B, Idris A et al (2015) Production of ultra-high concentration calcium alginate beads with prolonged dissolution profile. RSC Adv 5:36687–36695. https://doi.org/10.1039/C5RA03862F
Pereira R, Tojeira A, Vaz DC et al (2011) Preparation and characterization of films based on alginate and aloe vera. Int J Polym Anal Charact 16:449–464. https://doi.org/10.1080/1023666X.2011.599923
Fertah M, Belfkira A, Dahmanemontassir E et al (2017) Extraction and characterization of sodium alginate from Moroccan Laminaria digitata brown seaweed. Arab J Chem 10:S3707–S3714. https://doi.org/10.1016/j.arabjc.2014.05.003
Lawrie G, Keen I, Drew B et al (2007) Interactions between alginate and chitosan biopolymers characterized using FTIR and XPS. Biomacromol 8:2533–2541. https://doi.org/10.1021/bm070014y
Costa MJ, Marques AM, Pastrana LM et al (2018) Physicochemical properties of alginate-based films: effect of ionic crosslinking and mannuronic and guluronic acid ratio. Food Hydrocoll 81:442–448. https://doi.org/10.1016/j.foodhyd.2018.03.014
Liu S, Li Y, Li L (2017) Enhanced stability and mechanical strength of sodium alginate composite films. Carbohydr Polym 160:62–70. https://doi.org/10.1016/j.carbpol.2016.12.048
Sadeghi M, Hosseinzadeh H (2013) Synthesis and properties of collagen-g-poly(sodium acrylate-co-2-hydroxyethylacrylate) superabsorbent hydrogels. Brazilian J Chem Eng 30:379–389. https://doi.org/10.1590/S0104-66322013000200015
Marangoni Júnior L, Rodrigues PR, da Silva RG et al (2022) Improving the mechanical properties and thermal stability of sodium alginate/hydrolyzed collagen films through the incorporation of SiO2. Curr Res Food Sci 5:96–101. https://doi.org/10.1016/j.crfs.2021.12.012
Aloui H, Deshmukh AR, Khomlaem C, Kim BS (2021) Novel composite films based on sodium alginate and gallnut extract with enhanced antioxidant, antimicrobial, barrier and mechanical properties. Food Hydrocoll 113:106508. https://doi.org/10.1016/j.foodhyd.2020.106508
Zhang R, Guo J, Liu Y et al (2018) Effects of sodium salt types on the intermolecular interaction of sodium alginate/antarctic krill protein composite fibers. Carbohydr Polym 189:72–78. https://doi.org/10.1016/j.carbpol.2018.02.013
Helmiyati AM (2017) Characterization and properties of sodium alginate from brown algae used as an ecofriendly superabsorbent. IOP Conf Ser Mater Sci Eng 188:012019. https://doi.org/10.1088/1757-899X/188/1/012019
Pastor C, Sánchez-González L, Cháfer M et al (2010) Physical and antifungal properties of hydroxypropylmethylcellulose based films containing propolis as affected by moisture content. Carbohydr Polym 82:1174–1183. https://doi.org/10.1016/j.carbpol.2010.06.051
Xue F, Zhao M, Liu X et al (2021) Physicochemical properties of chitosan/zein/essential oil emulsion-based active films functionalized by polyphenols. Futur Foods 3:100033. https://doi.org/10.1016/j.fufo.2021.100033
Roy S, Kim HC, Kim JW et al (2020) Incorporation of melanin nanoparticles improves UV-shielding, mechanical and antioxidant properties of cellulose nanofiber based nanocomposite films. Mater Today Commun 24:100984. https://doi.org/10.1016/j.mtcomm.2020.100984
Roy S, Rhim J-W (2021) Preparation of gelatin/carrageenan-based color-indicator film integrated with Shikonin and Propolis for smart food packaging applications. ACS Appl Bio Mater 4:770–779. https://doi.org/10.1021/acsabm.0c01353
Hajinezhad S, Razavizadeh BM, Niazmand R (2020) Study of antimicrobial and physicochemical properties of LDPE/propolis extruded films. Polym Bull 77:4335–4353. https://doi.org/10.1007/s00289-019-02965-y
Khodayari M, Basti AA, Khanjari A et al (2019) Effect of poly(lactic acid) films incorporated with different concentrations of Tanacetum balsamita essential oil, propolis ethanolic extract and cellulose nanocrystals on shelf life extension of vacuum-packed cooked sausages. Food Packag Shelf Life 19:200–209. https://doi.org/10.1016/j.fpsl.2018.11.009
Marangoni Júnior L, Jamróz E, de Gonçalves S, Á, et al (2022) Preparation and characterization of sodium alginate films with propolis extract and nano-SiO2. Food Hydrocoll Heal 2:100094. https://doi.org/10.1016/j.fhfh.2022.100094
Brunauer S, Deming LS, Deming WE, Teller E (1940) On a theory of the van der waals adsorption of gases. J Am Chem Soc 62:1723–1732. https://doi.org/10.1021/ja01864a025
Smith SE (1947) The sorption of water vapor by high polymers. J Am Chem Soc 69:646–651. https://doi.org/10.1021/ja01195a053
Nazreen AZ, Jai J, Abdulbari Ali S, Mohamed Manshor N (2020) Moisture adsorption isotherm model for edible food film packaging – a review. Sci Res J 17:221. https://doi.org/10.24191/srj.v17i2.10160
Ahmad M, Nirmal NP, Danish M et al (2016) Characterisation of composite films fabricated from collagen/chitosan and collagen/soy protein isolate for food packaging applications. RSC Adv 6:82191–82204. https://doi.org/10.1039/c6ra13043g
Fakhreddin Hosseini S, Rezaei M, Zandi M, Ghavi FF (2013) Preparation and functional properties of fish gelatin-chitosan blend edible films. Food Chem 136:1490–1495. https://doi.org/10.1016/j.foodchem.2012.09.081
Guerrero P, Nur Hanani ZA, Kerry JP, De La Caba K (2011) Characterization of soy protein-based films prepared with acids and oils by compression. J Food Eng 107:41–49. https://doi.org/10.1016/j.jfoodeng.2011.06.003
Jridi M, Hajji S, Ben AH et al (2014) Physical, structural, antioxidant and antimicrobial properties of gelatin-chitosan composite edible films. Int J Biol Macromol 67:373–379. https://doi.org/10.1016/j.ijbiomac.2014.03.054
Kowalonek J, Kaczmarek H (2010) Studies of pectin/polyvinylpyrrolidone blends exposed to ultraviolet radiation. Eur Polym J 46:345–353. https://doi.org/10.1016/j.eurpolymj.2009.09.015
Gomaa M, Hifney AF, Fawzy MA, Abdel-Gawad KM (2018) Use of seaweed and filamentous fungus derived polysaccharides in the development of alginate-chitosan edible films containing fucoidan: Study of moisture sorption, polyphenol release and antioxidant properties. Food Hydrocoll 82:239–247. https://doi.org/10.1016/j.foodhyd.2018.03.056
Borges JG, Silva AG, Cervi-Bitencourt CM et al (2016) Lecithin, gelatin and hydrolyzed collagen orally disintegrating films: Functional properties. Int J Biol Macromol 86:907–916. https://doi.org/10.1016/j.ijbiomac.2016.01.089
Arruda C, Pena Ribeiro V, Oliveira Almeida M et al (2020) Effect of light, oxygen and temperature on the stability of artepillin C and p-coumaric acid from Brazilian green propolis. J Pharm Biomed Anal 178:112922. https://doi.org/10.1016/j.jpba.2019.112922
Andrade JKS, Denadai M, de Oliveira CS et al (2017) Evaluation of bioactive compounds potential and antioxidant activity of brown, green and red propolis from Brazilian northeast region. Food Res Int 101:129–138. https://doi.org/10.1016/j.foodres.2017.08.066
Acknowledgements
The authors acknowledge the National Council for Scientific and Technological Development (CNPq), Grant Number 403595/2021-5; and the São Paulo Research Foundation (FAPESP) for the post-doctoral fellowship of L. Marangoni Júnior (2021/04043-2). This study was partly financed by the Coordination for the Improvement of Higher Education Personnel – Brazil (CAPES) – Financial Code 001.
Funding
This study was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo, (Grant Number 2021/04043-2), Conselho Nacional de Desenvolvimento Científico e Tecnológico, (Grant Number 403595/2021-5).
Author information
Authors and Affiliations
Contributions
LMJ: Conceptualization, Data curation, Formal analysis, Investigation, Writing – original draft. PRR: Data curation, Formal analysis, Investigation, Writing – original draft.EJ: Data curation, Investigation, Writing – original draft. RGdaS: Formal analysis, Writing – review & editing. RMVA: Formal analysis, Writing – review & editing. RPV: Conceptualization, Data curation, Funding acquisition, Investigation, Writing – original draft, Writing – review & editing.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Marangoni Júnior, L., Rodrigues, P.R., Jamróz, E. et al. Green Propolis Extract as an Antioxidant Additive for Active Films Based on Sodium Alginate and Hydrolyzed Collagen. J Polym Environ 31, 1853–1865 (2023). https://doi.org/10.1007/s10924-022-02722-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10924-022-02722-9