
Abstract
The sand cat, Felis margarita Loche, 1858, is a small desert cat with a fragmented distribution across the Sahara, Arabian Peninsula, and Central Asia. It is currently listed as Least Concern by the IUCN; however, its status in many countries is unknown. Sand cats are generally classified into four subspecies: F. m. margarita (North Africa and Sahara), F. m. harrisoni (Arabia), F. m. thinobia (Central Asia), and F. m. scheffeli (Pakistan). The aim of this study was to determine the validity of these subspecies using genetic analysis. Sequences from the mitochondrial control region, mitochondrial NADH subunit 5, and cytochrome b genes (643 bp) were generated in a sample set of 47 animals of known geographical origin, from across the sand cat’s range over the past 100 years. The results of the analysis suggest some degree of genetic differentiation between the African populations (F. m. margarita) and those of Arabian or Central Asian origin, which merits further investigation with nuclear loci and further sampling of intermediate geographical locations. There is little genetic justification for differentiation of the other proposed subspecies (F. m. harrisoni, F. m. thinobia, and F. m. scheffeli), which differ only by 1–3 bp mutations in their haplotypes. The genetic diversity of a set of 86 captive sand cat samples is also generated and compared to facilitate future conservation management of the species in captivity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
There are at least 26 different ways of defining species (Frankham et al. 2012) and countless other ways of defining groups below the level of the taxon (see for example Cronin et al. 2015). Although to some evolutionary biologists the definition of taxa can seem arbitrary given the evolutionary continuum between all living organisms, taxonomic classification can have a huge practical impact on conservation. Species and subspecies units are the currency of international agreements and legislation (e.g., IUCN red-list, CITES, and the US Endangered Species Act (ESA), Haig et al. 2006) and as such, nomenclature can impact the monitoring of illegal trade, captive breeding, translocation, and range re-establishment. Accepted taxonomic classifications, however, are often a historical relict based on the examination of small numbers of specimens, and as such, have the potential to misdirect conservation and management efforts. Such is the case for the sand cat.
The sand cat, Felis margarita Loche, 1858, is a small felid with a wide distribution in deserts from the Sahara to the Arabian Peninsula to southwest Asia. It is the only felid found in true desert (Sunquist and Sunquist 2002). Perhaps due to its elusive nature and presence in a harsh environment, sand cats are understudied and relatively little information is available regarding their distribution, ecology, or evolutionary history.
Genetic studies place sand cats within the domestic cat lineage of the Felidae family (Johnson et al. 2006). A recently derived group, the domestic cat lineage arose in Eurasia an estimated 6.2–6.7 million years ago (Mya) and also includes domestic cats (F. catus Linnaeus, 1758), their wild progenitors (F. silvestris Schreber, 1777) as well as other small felid species: the black footed cat (Felis nigripes Burchell, 1824), jungle cat (Felis chaus Schreber, 1777), and Chinese desert cat (Felis bieti Milne-Edwards, 1892, though it is unresolved if this is a subspecies of F. silvestris or a separate Felis species (Nowell and Jackson 1996)). Sand cats are closely related to F. silvestris, F. bieti, and F. catus (Driscoll et al. 2007), diverging away from this group an estimated 2.5 Mya (Johnson et al. 2006). Johnson et al. (2006) proposed subsequent migration of the sand cat, wildcat, and black footed cat from Asia into Africa during the Pleistocene era. There is some range overlap between sand cats and wildcats (subsp. F. silvestris lybica and F. silvestris ornata), which are present in a range of habitats including scrub and true desert (though in small numbers) (Nowell and Jackson 1996).
The sand cat’s range is thought to be discontinuous (Fig. 1), but it is unclear if this is for ecological reasons or due to lack of recording effort. Currently, the sand cat has been reported (from confirmed sightings or specimens) in Morocco, western Sahara, Algeria, Mauritania, Mali, Niger, Egypt (east of the river Nile), and the Sinai Peninsula into Jordan and Syria (Abbadi 1989; Sliwa et al. 2016). On the Arabian Peninsula there are confirmed records of the sand cat in Kuwait, Oman, Qatar, Saudi Arabia, and the United Arab Emirates (Banfield et al. 2014; Sliwa et al. 2016). There is one 50-year old record of a specimen collected in Yemen (Harrison and Bates 1991; Banfield et al. 2014). In southwest Asia it is reported in Iran, Turkmenistan, Uzbekistan, and Kazakhstan, and it was also reported in the Baluchistan province of Pakistan (Sliwa 2013; Sliwa et al. 2016). The recorded distribution of the sand cat may expand as further studies are carried out. For example, sand cats have recently been recorded for the first time in Iraq (Mohammad et al. 2013) and Chad (Rabeil et al. 2016), and for the first time in 10 years in the United Arab Emirates (Ahmed et al. 2016).

Current accepted distribution for the sand cat (IUCN). Gaps in distribution may also represent data gaps. On the map are the locations of the four main proposed subspecies of sand cat: F. m. margarita, F. m. harrisoni, F. m. scheffeli, and F. m. thinobia (Adapted from: www.maps.iucnredlist.org)
The species is currently listed as Least Concern on the IUCN Red List, though this is based on a current lack of reliable population estimates and may change as new information arises (Sliwa et al. 2016). The population in Pakistan was heavily exploited for the zoo trade to Europe and the US during the 1960s and 1970s, and faced habitat destruction during the 1990s (Sausman 1997; Sliwa et al. 2016). There is concern that the sand cat may already be extinct, or close to extinction, in the region (Sliwa et al. 2016). A lack of recent recorded sightings or specimens from Yemen or Israel also raises concerns about local extinctions of these populations.
Taxonomic History
Owing to apparently fragmented distribution and infrequent sampling, taxonomic classification of the sand cat has been fairly haphazard, with a number of taxa described during the twentieth century.
Sand cats were first described from North Africa by Victor Loche in 1858 (Loche 1858). In 1926, a Russian mammalogist, S. I. Ognev, described a similar desert cat, but from the opposite end of the sand cat range, from the Repetek Desert Reserve in Turkmenistan (Ognev 1926). Ognev placed his specimen in a separate genus, naming it Eremaelurus thinobius. Pocock (1938a, b) recognized two Saharan subspecies of F. margarita (based on single specimens): F. m. airensis (from In-Abbangarit west of Aïr, Central Niger) and F. m. meinertzhageni (El Golea, latitude 30o N in the Algerian Sahara), but these were later collapsed into a single African subspecies, F. m. margarita, by Hemmer et al. (1976). By the mid-twentieth century specimens had also been collected from the Empty Quarter of the Arabian Peninsula by W. P. Thesiger (Hemmer 1974a) and from Nushki, in Pakistan, by J. Anderson (Hemmer 1974b). These were described as two further subspecies: F. m. harrisoni from Arabia (Hemmer 1974a) and F. m. scheffeli from Pakistan (Hemmer 1974b). Heptner and Sludskii (1972) recognized only two subspecies of F. margarita: thinobia (Central Asia) and margarita (North Africa and Arabia).
Major revisions were made to the taxonomy during the 1970s, with Hemmer et al. (1976), consolidating all desert cats into a single species, F. margarita, with four subspecies: F. m. margarita, F. m. harrisoni, F. m. thinobia and F. m. scheffeli (Fig. 1), finally linking Loche’s and Ognev’s cats. This remains the accepted sand cat taxonomy. Classification was based on morphological differences, specifically pelage coloration and markings (Supplementary Fig. 1), skull size and proportions, and apparent geographical separation. Hemmer (1974a) stated that western cats (i.e., North African) are generally brighter in color and have distinct stripes, whereas eastern specimens are paler with less distinct markings, with Arabian cats intermediate between the two. However, it should be noted that an animal’s age is also likely to be an important factor in the strength of markings. Hemmer (1974b) also noted that Pakistani and Arabian cats have more rings on their tails, and that Arabian cats have the smallest skulls. Schauenberg (1974), in comparison, actually noted a decrease in skull size from east to west, with western cats having the smallest skulls.
No phylogenetic analysis of in situ sand cats has been carried out to date and such an analysis is listed as a priority for the species (Sliwa et al. 2016; Kitchener et al. 2017). Unresolved taxonomy may be a barrier to future conservation management and hence its resolution is vital prior to decisions about population reinforcement or reintroduction as well as ex situ population management for conservation purposes. Although a number of considerations aside from genetic data should contribute to decisions regarding taxonomic re-classification (e.g., Kitchener et al. 2017), molecular data are nevertheless an extremely valuable component of this assessment. Here, we use data from three mitochondrial genes (control region, NADH subunit 5, and cytochrome b) to understand whether the current taxonomy of the sand cat is supported from a molecular genetic perspective.
Materials and Methods
Sample Collection
Samples for this study came from a mixture of 199 captive, wild, and historical specimens of known geographical origin, with individuals from across the sand cat’s range. Seventy-eight historical samples were collected from institutions across Europe (Zoological Institute of the Russian Academy of Sciences, St. Petersburg; Muséum National d’Histoire Naturelle, Paris; Harrison Institute, Sevenoaks; and the Senckenberg Museum, Frankfurt) and consisted of small pieces of skin, footpad, cartilage, skull tissue and/or turbinate bones. Thirty-five samples were collected from wild individuals, including eight wild-born, captive animals. A further 86 samples came from individuals born in captivity, reported to be of the Arabian subspecies, but ascribed an unknown origin. These samples were used to assess the genetic structure and diversity of the captive population. Samples not of historic origin consisted of hair, blood, and tissue (see Supplementary Material for a full list of samples).
DNA Extraction
The historical samples were extracted using QIAmp DNA Investigator Kit (Qiagen), according to the manufacturer’s instructions, with some samples requiring a prolonged digest (up to 21 h). All samples were extracted in sterile conditions and isolated from downstream PCR processes. Extractions were carried out in small batches (6–8) and separately from contemporary samples to minimize the risk of cross-contamination.
The modern samples were extracted using DNeasy Blood & Tissue Kit (Qiagen) as per the manufacturer’s instructions. Extraction and PCR was conducted once processing and analysis of the historical samples had concluded.
DNA Amplification and Sequencing
Fragments of the mitochondrial control region (d-loop; located in the hypervariable region 1), cytochrome b (cytB; located at the 3′ end of the gene and 50 bp into tRNA-thr), and NADH dehydrogenase 5 (ND5; located towards the 5′ end) were amplified using the primer combinations shown in Table 1. Three overlapping cytochrome b primers were used to generate a 400 bp cytochrome b fragment if the long primer pairs would not amplify.
PCR was carried out using 2 μl (1–10 ng/μl) template DNA, 14 μl Maxima Hot Start PCR Master Mix (Thermo Fisher), 2 μl of forward primer and 2 μl of reverse (10 μM). The following PCR program was used: 5 min initial denaturation at 95 °C followed by 40 cycles of 1 min denaturation (95 °C), 1 min primer annealing at 55 °C, and 1 min elongation (72 °C) ending with a further 5 min extension at 72 °C. A negative control (Master Mix and primers only) was used in every PCR, although a positive control was excluded to prevent cross-contamination.
Following PCR, the fragments were visualized on a 1.5% agarose gel; successfully amplified samples were cleaned up using 0.5 μl of each EXO1 and FastAP and incubated at 37 °C for 45 min and 15 min at 80 °C. 2 μl of cleaned up PCR product was sequenced in both directions using BigDye Terminator Kit (Applied Biosystems) as per the manufacturer’s instructions.
Sequence Analysis
Sequences were trimmed and aligned using GENEIOUS 8.0 (www.geneious.com, Kearse et al. 2012). Sequences were trimmed to 90 bp (d-loop), 384 bp (cytB), and 169 bp (ND5). A concatenated sequence of all three genes was generated for each individual (643 bp), using the mitochondrial genome of a domestic cat as a reference (NCBI KP202278). All mitochondrial DNA sequences were deposited in GenBank (accession MK606117–606132).
Haplotype frequency, population pairwise Fst, haplotype and nucleotide diversity, neutrality tests (Tajima’s D and Fu’s FS), and an AMOVA (10,000 permutations) were calculated in ARLEQUIN 3.5 (Excoffier and Lischer 2010) and median-joining haplotype networks were calculated and drawn in PopArt v1.7 (www.popart.otago.ac.nz).
Neighbor-joining, maximum-likelihood, and Bayesian inference trees were inferred in GENEIOUS, with F. silvestris lybica and Panthera leo as outgroups (NCBI KP202275 and KF907306). Neighbor-joining trees were inferred using the Tamura-Nei distance model with 100 bootstrap iterations and 75% support threshold. Maximum-likelihood trees were calculated using PhyML, using the best fitting model, Hasegawa-Kishino-Yano (HKY, Hasegawa et al. 1985) with a proportion of invariable sites of 0.9, as estimated by MODELTEST (Posada and Crandall 1998). These trees were also calculated with 100 bootstrap iterations. Bayesian trees were inferred using the MrBayes plugin to GENEIOUS 8.0, using HKY85 model of evolution, MCMC chain length of 1,100,000, and burn-in of 100,000. For each tree-building method, four sets of trees were produced; one for the whole 643 bp concatenated fragment and one each for d-loop, cytB, and ND5 gene fragments.
Divergence times were estimated using RevBayes (Höhna et al. 2014, 2016). Felis silvestris (KP202275) was included as an outgroup and the divergence time between F. silvestris and F. margarita (as estimated by Johnson et al. 2006) was used to calibrate the tree. A Rev. file was created with the following parameters: HKY + G model of substitution, constant birth-death rate as a tree prior, and a global molecular clock. The estimated divergence time between wildcats and sand cats was placed on the root of the tree with a normal distribution (mean = 2.49 Mya, min = 1.72, max = 3.67). We ran MCMC chains of length 50,000, sampling every ten steps, with a 10% burn-in, and MCMC chain convergence and effective sample sizes were assessed using TRACER v1.6. The annotated maximum a posteriori tree was built in RevBayes and viewed in FigTree v1.4.3; node ages and 95% HPDs are estimated from this tree.
Data Availability Statement
All sequence data are made available online via NCBI GenBank (for accession numbers see above).
Results
Sample Success
Of the 199 samples examined, 71 failed to amplify and a further eight were excluded due to incomplete data (though returned NCBI BLAST hits to sand cat). Four additional samples (SCA118, 179, 180, and 181) amplified but returned as wildcat (F. silvestris). SCA118 is a skull sample and therefore the mismatch could be the result of mislabelling or misidentification at the time of its collection. SCA179–181 were three modern samples from Mauritania, all of which were collected from road-kill specimens where species identification was not straightforward (as can be seen in Supplementary Fig. 2).
The dataset used for phylogenetic analyses consisted of sequences from 116 historic and modern samples that amplified at all three gene regions, 46 of which were wild individuals, including wild-born individuals now in captivity (Supplementary Table 1).
Genetic Diversity
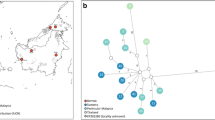
Eleven d-loop, seven cytB, and six ND5 haplotypes were identified across the entire dataset of 116 individuals. The concatenated sequences gave 16 unique haplotypes (Fig. 2) and were named AB to AR. A single haplotype (AD) matched the pre-existing NCBI sequence KP202276 (geographical origin unknown). In the wild dataset, the highest number of haplotypes (n = 5) was found in the Arabian samples and three haplotypes were found in Pakistan, Turkmenistan, and Africa, respectively (Table 2). Haplotype diversity was highest in the samples from Pakistan, followed by Arabia. Nucleotide diversity was highest in the samples from Turkmenistan, followed by Pakistan. The lowest haplotype and nucleotide diversities were found in the African samples, where three closely related haplotypes were identified. Haplotype (AB) was present in the vast majority of African samples (Table 2).

A Median-Joining (MJ) networks showing phylogeographic structure in the major populations of sand cat globally. The network is based on a fragment of ND5, d-loop, and cytB mtDNA genes that totals 643 bp in length. Each ball represents a haplotype, color coded by origin. The number of black tick marks between haplotypes represents the number of single base pair mutations separating them. a: individuals of wild origin only, b: all wild and captive cats sampled. Haplotype AD is also identical to a sequence of an unknown origin captive cat submitted to NCBI Genbank (KP202276, Li et al. 2016). Two haplotypes (AE and AL) have only been found within captivity. c: Single nucleotide polymorphisms in the fragment mapped against KP202275
Analysis of between- and within-population genetic variation, AMOVA, revealed that the majority of genetic variation (84.74%) was captured among populations, with 12.26% within populations (Fst = 0.877, p < 0.001, d.f. = 45). No haplotypes were shared between the four geographic sampling locations. Pairwise Fst was greatest between Africa and Pakistan and Africa and Turkmenistan and least between Turkmenistan and Pakistan and Turkmenistan and Arabia (Table 4).
Six haplotypes were recovered within the 66 captive-born individuals, of which two (AE and AL) were not found within the wild samples. Haplotype diversity in captivity was 0.7520 and nucleotide diversity was 0.003227, compared to 0.7372 and 0.006688 in the wild samples (Table 4).
Demography
The results of the Tajima’s D (Tajima 1989) and Fu’s FS (Fu 1997) neutrality tests are included in Table 3. Significant values are indicated; Fu’s FS values are significant at the 5% level if the p value is below 0.02. Significant D or FS values are signatures of a departure from the neutral model of evolution due to selection or demographic expansion/contraction, and this is observed in the Arabian sand cat population. In this population, both Tajima’s D and Fu’s FS are significantly negative, evidence of a selective sweep or recent demographic expansion.
Tree-Building
At d-loop, tree building revealed no statistically supported phylogenetic sub-structure within the sand cat dataset, using both neighbor-joining (NJ) and maximum-likelihood (ML) methods. Using Bayesian inference, a cluster of haplotypes AB, AQ, and AR was supported (moderately) with a posterior probability of 88.5%. At ND5 no tree building method supported any sub-structure in the data. At cytB, all three methods of tree building supported the separation of the cytB sequences associated with haplotypes AB, AQ, and AR with 99/100% bootstrap and 89% posterior probability.
The concatenated three-gene fragment (643 bp) revealed NJ, ML, and Bayesian trees with concordant structures; all methods supported the separation of haplotypes AB, AQ, and AR (100% posterior probability and 99% and 100% bootstrap support for ML and NJ, respectively, see Fig. 3). No other phylogenetic sub-structure was statistically supported. The haplotypes AB, AQ, and AR were all found in samples of animals from Africa.

Tree at concatenated 634 bp region of d-loop, cytB, and ND5. Statistical support for Bayesian/Maximum-likelihood and Neighbor-Joining methods is shown
Divergence Time Estimate
The root node was estimated at 2.63 Mya (95% HPD: 1.72–3.50) and the divergence time between the African sand cat clade, F. margarita margarita, and the rest of the population was estimated to be 1.72 Mya 95% HPD:0.82–2.74) (Supplementary Fig. 1).
Haplotype Networks
The median-joining network of sand cat haplotypes from the wild (Fig. 2a) recovered a pattern of genetic relatedness roughly corresponding with geography. There was a lack of haplotype sharing between sampled geographical locations, but also a relatively low degree of genetic divergence, with 1–7 bp mutations between adjacent haplotypes. Average numbers of pairwise (bp mutation) differences were greatest between African and Pakistan (7.91) and least between Turkmenistan and Arabia (3.78). Average numbers of pairwise (bp mutation) differences within populations were highest in Turkmenistan (2.57) and lowest in Africa (0.22) (Table 4). The maximum divergence at cytochrome b haplotypes was 4 bp corresponding to 1.04% divergence.
Addition of the captive samples to the network (Fig. 2b) revealed that one of the two haplotypes found only in captivity (AE) was positioned at an inferred node within the wild-only network (compare Fig. 2a-b).
Discussion
This study investigated the relatedness of the four commonly recognized sand cat subspecies as proposed by Hemmer et al. (1976). Our results give provisional evidence to support the F. m. margarita subspecies grouping; individuals of African origin appear to be genetically distinct from those in the rest of the range (Fig. 2, Table 4). A minimum 6 bp difference between F. m. margarita and the other putative subspecies resulted in a monophyletic clade of African individuals, statistically supported across three phylogenetic tree-building methods (Fig. 3). Division was underpinned by a 3–4 bp difference at cytochrome b between F. m. margarita and the most geographically proximate subspecies, Arabian F. m. harrisoni. This equates to 0.78–1.04% divergence at this gene, a value considered at the lower end of the scale for subspecies divergence (Baker and Bradley 2006). It is possible that the results presented here represent a split between African sand cats and those of Arabia and Central Asia. A pattern of subspecies divergence either side of the Sinai Peninsula has been reported in two other carnivoran species: the cheetah, Acinonyx jubatus, and the African golden wolf/golden jackal, Canis sp. (Charruau et al. 2011; Koepfli et al. 2015). The Sinai Peninsula as a barrier to gene flow between sand cat populations may act as a possible explanation for the population differentiation observed here.
Divergence of the African sand cat clade was calculated to be around 1.72 Mya. However, the 95% highest posterior density interval was large (0.82–2.74 Mya) and overlapping with the estimated divergence date of the sand cat species within the domestic cat lineage, so is not helpful to examine the evolutionary history of this putative subspecies. Further studies including whole genome mtDNA and nuclear sequencing would be useful here (e.g., Li et al. 2016).
Despite genetic evidence to support the subspecies group F. m. margarita, we would suggest that as divergence is limited, and a number of intermediate sand cat populations have not been sampled (notably central Iran, southern Egypt, and the Sinai Peninsula), this should only be viewed as tentative. When defining taxa for conservation purposes, it is important to take multiple lines of evidence into account. In general, divergent monophyletic clades at mtDNA genes (particular cytochrome b) should be viewed as a necessary, but not a sufficient, character for the delineation of mammalian taxa (Frankham et al. 2011, 2012; Kitchener et al. 2017). It is entirely possible that the disjunction between Africa and other populations may simply reflect a disjunction in sampling.
Another notable feature of the sampled F. m. margarita population was low genetic variation (Table 3). The 26 samples examined were collected in Sudan, Algeria, Mauritania, and Morocco (western Saharan region), with collection spanning 1930–2014. Although this sampling is far from comprehensive, the data nevertheless seem indicative of low overall genetic diversity in the African population.
Range expansion generally involves a series of population bottlenecks and founder effects, resulting in decreasing genetic diversity at increasing distances from the source population (Austerlitz et al. 1997; Excoffier et al. 2009). This has been reported in other small mammals using cytochrome b and mitochondrial control region genes, for example, late Pleistocene migration of the Egyptian mongoose from North Africa to the Iberian Peninsula (Barros et al. 2016). It seems likely that reduced genetic diversity in F. m. margarita is a signature of recent colonization of Africa from Asia, and corresponds to the late Pleistocene migration of F. margarita proposed by Johnson et al. (2006). However, recent population contraction was not supported by the results of the neutrality tests (Table 3). Neither Tajima’s D or Fu’s FS were significant in the African sand cat population, indicative of a stable population. Assessment of genetic variation within F. silvestris lybica or F. nigripes, which are thought to have migrated from Asia to Africa around the same time (Johnson et al. 2006), would potentially offer an informative comparison to the sand cat.
In the other sand cat populations, F. m. harrisoni, F. m. thinobia, and F. m. scheffeli, there is some correspondence between haplotype divergence and geography, but we would suggest this is not significant enough to merit their recognition as separate subspecies. This is particularly true of F. m. thinobia and F. m. scheffeli, where some haplotypes are separated by only 1 bp difference (Fig. 2). On average, pairwise differentiation between the groups is 4.22 bp (Table 4) and the arrangement of the groups is not monophyletic. The minimum division between the Arabian population and those from Pakistan and Turkmenistan is 3 bp, although the relationship may be closer if it is assumed that haplotypes AE originated in Arabia, which is most likely given historical records. Genetic diversity is higher in these populations (Table 3) and comparable to that found in previous study of the ex situ sand cat population (Witzenberger and Hochkirch 2013). For the Arabian population both Tajima’s D and Fu’s F were significantly negative (Table 3); this indicates deviation from neutrality and may be evidence of recent population expansion.
We also examined the genetic structure and diversity in the ex situ sand cat population. The current captive population consists only of the putative subspecies F. m. harrisoni. However, F. m. scheffeli, from Pakistan, were kept from the 1970s until fairly recently within the American Zoo Association population, so an origin from either geographic location cannot be ruled out. As of 2016, 156 sand cats were represented in 41 North American, Eurasian, and Middle Eastern zoos and institutions (n = 12, 22, and 7, respectively) (Breton et al. 2016). Global coordination of breeding began in 1988 and studbooks are maintained for this species by the Arabian and European Zoo and Aquarium Associations and the American Zoo Association (Breton 2017). Previous genetic assessment of the captive population (the samples of which were also included in the study) revealed that the captive population has arisen from at least three genetic lineages. These lineages did not correspond to geographical origin, in the cases where it was known, indicating they most likely represent founders or families of founders (Witzenberger and Hochkirch 2013). Our results found six mitochondrial haplotypes in the captive population (Table 2), confirming that at least six female founders are likely to have established the population. A relatively high level of genetic diversity in the captive population was found by Witzenberger and Hochkirch (2013) and this finding is also recovered by this study. Levels of genetic diversity are roughly equivalent to those found within each of the wild populations (Table 3), suggesting that the diversity within captivity is at an acceptable level. However, a larger sample size of each population in the wild and the addition of nuclear markers would be needed to benchmark this fully.
In conclusion, we find tentative evidence to support the subspecies grouping of the African sand cat, F. m. margarita, but limited divergence between the three remaining subspecies groups, F. m. harrisoni, F. m. thinobia, and F. m. scheffeli. This finding may be useful evidence when considering the validity of these subspecies to accurately inform species management and conservation.
References
Abbadi M (1989) Radiotelemetric observations on sand cats (Felis margarita) in the Aravah Valley. Isr J Zool 36: 155–156
Ahmed S, Al Zaabi R, Soorae P, Shah JN, Al Hammadi E, Pusey R, Al Dhaheri S (2016) Rediscovering the Arabian sand cat (Felis margarita harrisoni) after a gap of 10 years using camera traps in the western region of Abu Dhabi, United Arab Emirates. Eur J Wildlife Res 62:627–631
Austerlitz F, Jung-Muller B, Godelle B, Gouyon PH (1997) Evolution of coalescence times, genetic diversity and structure during colonization. Theor Popul Biol 51:148–164
Baker RJ, Bradley RD (2006) Speciation in mammals and the genetic species concept. J Mammal 87:643–662
Banfield LM, al Qahtani H, Mallon D (2014) Arabian Sand Cat Felis margarita harrisoni Status Review and Conservation Strategy. Al Ain Zoo, Al Ain
Barros T, Gaubert P, Rocha RG, Bandeira V, Souto L, Mira A, Carlos F (2016) Mitochondrial demographic history of Egyptian mongoose (Herpestes ichneumon), an expanding carnivore in the Iberian Peninsula. Mammal Biol 81:176–184
Breton G (2017) International Studbook for the Sand Cat (Felis margarita). Volume XXIII
Breton G, Sliwa A, Azizi S, Essalhi A (2016) Sand cats in the Moroccan Sahara, preliminary results of a new study. Cat News 63:7–10
Charruau P, Fernandes C, Orozco-terWengel P, Peters J, Hunter L, Ziaie H, Jourabchian A, Jowkar H, Schaller G, Ostrowski S, Vercammen P, Grange T, Schlötterer C, Kotze A, Geigl EM, Walzer C, Burger PA (2011) Phylogeography, genetic structure and population divergence time of cheetahs in Africa and Asia: evidence for long-term geographic isolates. Mol Ecol 20:706–724
Cronin MA, Cánovas A, Bannasch DL, Oberbauer AM, Medrano JF (2015) Wolf subspecies: reply to Weckworth et al. and Fredrickson et al. J Hered 106: 417–419
Driscoll CA, Menotti-Raymond M, Roca AL, Hupe K, Johnson WE, Geffen E, Harley EH, Delibes M, Pontier D, Kitchener AC, Yamaguchi N, O’Brien SJ, Macdonald DW (2007) The Near Eastern origin of cat domestication. Science 317: 519–523
Excoffier L, Lischer HE (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and windows. Mol Ecol Resour 10:564–567
Excoffier L, Foll M, Petit RJ (2009) Genetic consequences of range expansions. Annu Rev Ecol Evol Syst 40:481–501
Frankham R, Ballou JD, Dudash MR, Eldridge MDB, Fenster CB, Lacy RC, Mendelson III JR, Porton IJ, Ralls K, Ryder OA (2012) Implications of different species concepts for conserving biodiversity. Biol Cons 153:25–31
Frankham R, Ballou JD, Eldridge MDB, Lacy RC, Ralls K, Dudash MR, Fenster CB (2011) Predicting the probability of outbreeding depression. Conserv Biol 25: 465–475
Fu YX (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147:915–925
Haig SM, Beever EA, Chambers SM, Draheim HM, Dugger BD, Dunham S, Elliott-Smith E, Fontaine JB, Kesler DC, Knaus BJ, Lopes IF, Loschl P, Mullins TD, Sheffield LM (2006) Taxonomic considerations in listing subspecies under the U.S. endangered species act. Conserv Biol 20:1584–1594
Harrison DL, Bates PJJ (1991) The Mammals of Arabia (2nd ed.). Harrison Zoological Museum Publication, Sevenoaks
Hasegawa M, Lida Y, Yano T, Takaiwa F, Iwabuchi M (1985) Phylogenetic relationships among eukaryotic kingdoms inferred from ribosomal RNA sequences. J Mol Evol 22:32–38
Hemmer H (1974a) Studies of the systematics and biology of the sand cat. Kölner Zoo 17:11–20
Hemmer H (1974b) Felis margarita scheffeli new subspecies a new sand cat subspecies from the Nushki Desert Pakistan Mammalia Carnivora Felidae. Senckenb Biol 55:29–34
Hemmer H, Grubb P, Groves CP (1976) Notes on the sand cat, Felis margarita Loche, 1858. Z Säugetierk 41: 286–303
Heptner VH, Sludskii AA (1972) Mammals of the Soviet Union. Volume II Part 2. Carnivore (Hyenas and Cats). Vysshaya Shkola, Moscow. English Translation 1992, E.J. Brill, Leiden
Höhna S, Heath TA, Boussau B, Landis MJ, Ronquist F, Huelsenbeck JP (2014) Probabilistic graphical model representation in phylogenetics. Syst Biol 63:753–771
Höhna S, Landis MJ, Heath TA, Boussau B, Lartillot N, Moore BR, Huelsenbeck JP, Ronquist F (2016) RevBayes: Bayesian phylogenetic inference using graphical models and an interactive model-specification language. Syst Biol 65:726–736
Johnson WE, Eizirik E, Pecon-Slattery J, Murphy WJ, Antunes A, Teeling E, O’Brien SJ (2006) The late Miocene radiation of modern Felidae: a genetic assessment. Science 311:73–77
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A (2012) Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649
Kitchener AC, Breitenmoser-Wursten CH, Eizirik E, Gentry A, Werdelin L, Wilting A, Yamaguchi N, Abramov AV, Christiansen P, Driscoll C, Duckworth JW, Johnson W, Luo SJ, Meijaard E, O’Donoghue P, Sanderson J, Seymour K, Bruford M, Groves C, Hoffmann M, Nowell K, Timmons Z, Tobe S (2017) A revised taxonomy of the Felidae. The final report of the Cat Classification Task Force of the IUCN/SSC Cat Specialist Group. Cat News Special Issue 11:1–80
Koepfli KP, Pollinger J, Godinho R, Robinson J, Lea A, Hendricks S, Schweizer RM, Thalmann O, Silva P, Fan Z, Yurchenko AA, Dobrynin P, Makunin A, Cahill JA, Shapiro B, Álvares F, Brito JC, Geffen E, Leonard JA, Helgen KM, Johnson WE, O'Brien SJ, Van Valkenburgh B, Wayne RK (2015) Genome-wide evidence reveals that African and Eurasian golden jackals are distinct species. Curr Biol 25:2158–2165
Li G, Davis BW, Eizirik E, Murphy WJ (2016) Phylogenomic evidence for ancient hybridization in the genomes of living cats (Felidae). Genome Res 26(1):1–11
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452
Loche V (1858) Description d'une nouvelle espèce de Chat par M. le Capitaine Loche. Revue et Magasin de Zoologie Pure et Appliquée 2:49–51
Mohammad KM, Lahony SR, Al-Rammahi HM (2013) First record of the sand cat, Felis margarita Loche, 1858 (Mammalia: Carnivora, Felidae) from Iraq. Zool Middle East 59:358-359ig
Nowell K, Jackson P (1996) Wild Cats: Status Survey and Conservation Plan. International Union for Conservation of Nature and Natural Resources, Gland
Ognev SI (1926) A new genus and species of cat from the Transcaspian region. Annals of the Museum of Zoology, Academy of Science, USSR 27: 356–362
Pocock RI (1938a) The Algerian sand cat (Felis margarita Loche). Proc Zool Soc Lond B108: 41–46
Pocock RI (1938b) A new race of the sand cat (Felis margarita). Ann Mag Nat Hist Series 11 1:472–476
Posada D, Crandall KA (1998) MODELTEST: testing the model of DNA substitution. Bioinformatics 14:817–818
Rabeil T, Wacher T, Newby J (2016) Sand cat sightings in Niger and Chad. Cat News 63:11–14
Sausman KA (1997) Sand cat, Felis margarita, a true desert species. Internatl Zoo Yearbk 35:78–81
Schauenberg P (1974) Données nouvelles sur le chat des sables Felis margarita Loche, 1858. Revue suisse Zool 81:949–969
Sliwa A (2013) Felis margarita Loche. In: Kingdon JS, Happold D, Butynski T, Hoffmann M, Happold M, Kalina J (eds) The Mammals of Africa. Volumes 1–6. Academic Press, Amsterdam, pp 199–202
Sliwa A, Ghadirian T, Appel A, Banfield L, Sher Shah M, Wacher T (2016) Felis margarita. The IUCN Red List of Threatened Species 2016: e.T8541A50651884
Sunquist M, Sunquist F (2002) Wildcats of the World. University of Chicago Press, London
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Witzenberger KA, Hochkirch A (2013) Evaluating ex situ conservation projects: genetic structure of the captive population of the Arabian sand cat. Mammal Biol 78:379–382
Acknowledgments
We thank Katrin Krohmann at the Senckenberg Forschungsinstitut and Malcolm Pearch at the Harrison Institute for their help in providing us with access to their collections samples. We also thank staff at Al Wabra Wildlife Preservation, Breeding Centre for Endangered Arabian Wildlife, Bahrain Zoo, Colchester Zoo, Ebeltoft Zoo, Krakow Zoo, Le Parc des Felins (Nesles), London Zoo, Novosibirsk Zoo, Osnabrück Zoo, SOS Care (USA), Twycross Zoo, and Wuppertal Zoo for provision of samples to the authors of this manuscript.
Funding
This work was funded by Al Ain Zoo and the Royal Zoological Society of Scotland. JCB is supported by Fundação para a Ciência e Tecnologia (contract IF/459/2013).
Author information
Authors and Affiliations
Contributions
Project conception, management and design: HS, LB, AK,; Writing: JHM, HS, AK, LB; Analysis: JHM, HS; Wet Lab Analysis: JHM and MG; Sample collection/fieldwork: AT, AA, MQ, JCB, SA, GB, AS, KW Critical Review: ALL.
Corresponding author
Electronic supplementary material
ESM 1
(DOCX 6212 kb)
Rights and permissions
About this article

Cite this article
Howard-McCombe, J., Banfield, L., Kitchener, A.C. et al. A Mitochondrial Phylogeny of the Sand Cat (Felis margarita Loche, 1858). J Mammal Evol 27, 525–534 (2020). https://doi.org/10.1007/s10914-019-09473-w
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10914-019-09473-w