
Abstract
The genus Ctenomys comprises approximately 70 recognized living species of subterranean rodents endemic to South America. Phylogenetic studies to date, based on mitochondrial DNA data, place 44 recognized species in eight species groups and provide evidence for a burst of speciation early in the history of the genus. Species from Argentina, Uruguay, Bolivia, and southern Brazil are well studied at the phylogenetic level. However, the taxonomic status of the species inhabiting midwest and northern Brazil remains poorly understood. In this study, we construct phylogenies based on maximum likelihood and Bayesian inference methods with cytochrome b gene haplotypes of Ctenomys from midwest and northern Brazil and with haplotypes representative of the genus Ctenomys to place the sampled haplotypes into a phylogenetic framework; we also evaluate skull geometric morphometrics data among sampling sites to assess whether skull morphology corroborates the phylogenetic patterns observed. The results show that the sampling sites used in this study are represented by two species, namely, Ctenomys bicolor, which is present in the state of Rondônia, and Ctenomys nattereri, which is present in Mato Grosso and Bolivia. The results also reveal two lineages of Ctenomys distinct from C. bicolor and C. nattereri, henceforth called Ctenomys sp. “xingu” and Ctenomys sp. “central.” Both the species and lineages share a most recent common ancestor with C. boliviensis and are part of the boliviensis species group.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The order Rodentia is the most speciose order of mammals, with approximately 2277 species described and distributed in 34 families (Patton et al. 2015). These mammals occur in almost every continent, except for Antarctica, occupying arboreal, aquatic, terrestrial, or subterranean habitats (Nowak 1999). Subterranean rodents of the genus Ctenomys Blainville, 1826, are characterized by a dependency on tunnel systems, leaving the tunnels only for foraging, mating, or dispersing; they are also generally solitary and territorial, with each individual occupying its own tunnel system (Reig et al. 1990; Lacey et al. 2000).
The most speciose family of subterranean rodents, Ctenomyidae, comprises one extant genus, Ctenomys, which is distributed throughout the southern half of the Neotropical region (Fig. 1a) and contains approximately 70 valid species based on morphological, karyotypic, and molecular data (Bidau 2015; Patton et al. 2015; Freitas 2016). In the first phylogenetic study of the genus based on molecular data, Lessa and Cook (1998) identified strong monophyly between Octodontidae and Ctenomyidae in a cytochrome b (cyt b) phylogeny as well as evidence of polytomies in basal nodes of the phylogeny for Ctenomys, indicating a rapid radiation in the early history of the genus. Other studies included species from Uruguay and southern Brazil (D’Elía et al. 1999), compared chromosomal rearrangements with phylogenetic structuring (Slamovits et al. 2001), and provided phylogenetic estimates based on nuclear noncoding DNA markers (Castillo et al. 2005). Parada et al. (2011) identified eight monophyletic species groups with strong to moderate statistical support and estimated that most of the species groups originated approximately 3 million years ago, an estimate that is supported by the minimum estimated age of the oldest fossil known for the genus (~3.5 mya; Verzi et al. 2009). Upham and Patterson (2012) estimated that the genus Ctenomys originated approximately 4.3 mya, based on a nuclear and a mitochondrial gene. Finally, Gardner et al. (2014), based on cyt b sequences, provided the most comprehensive phylogenetic study to date for the genus Ctenomys, including 44 recognized species, four of which were described as new Bolivian species.

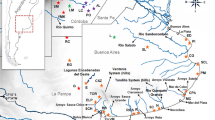
a Distribution of the genus Ctenomys throughout southern South America (light gray area). b Detailed distribution of sampled localities in the states of Mato Grosso (MT) and Rondônia (RO). Pimenta Bueno – PB, Pontes e Lacerda – PL, Cáceres – CA, Sapezal – SP, Nova Olímpia – NO, Tangará da Serra – TS, Nova Mutum – NM, Nova Ubiratã 1 – NU1, Nova Ubiratã 2 – NU2, Feliz Natal – FN
In Brazil, eight species of Ctenomys are currently known: five in southern Brazil (Rio Grande do Sul State – RS), namely, Ctenomys torquatus Lichtenstein, 1830, Ctenomys minutus Nehring, 1887, Ctenomys lami Freitas, 2001, Ctenomys flamarioni Travi, 1981, and Ctenomys ibicuiensis Freitas et al., 2012; one in the state of Mato Grosso (MT), namely, Ctenomys nattereri Wagner, 1848; and two species, namely, Ctenomys rondoni Miranda Ribeiro, 1914, and Ctenomys bicolor Miranda Ribeiro, 1914, in the state of Rondônia (RO) (Bidau and Ávila-Pires 2009). While species from southern Brazil, Argentina, Uruguay, and Bolivia are considerably well studied at the phylogenetic level (Lessa and Cook 1998; D’Elía et al. 1999; Slamovits et al. 2001; Castillo et al. 2005; Parada et al. 2011; Freitas et al. 2012; Gardner et al. 2014), the taxonomic status for the genus Ctenomys in midwest and northern Brazil remains poorly resolved. A revalidation of C. bicolor as a species based on molecular, karyotypic and morphological evidence (Stolz et al. 2013) is the only study published to date that involves individuals from these regions of Brazil. Considering the species distributed in midwest and northern Brazil, C. bicolor and C. nattereri are included in the boliviensis species group and are sister taxa to the Bolivian species Ctenomys boliviensis Waterhouse, 1848, and Ctenomys steinbachi Thomas, 1907 (Parada et al. 2011; Stolz et al. 2013; Gardner et al. 2014).
Recently, we sampled new sampling sites for Ctenomys in midwest and northern Brazil, further expanding the known distribution for the genus towards the Brazilian Amazon forest. Indeed, some of the individuals we sampled in the northernmost sampling sites were located in forests in areas of transition between the Amazonia and Cerrado (savanna) biomes of Brazil (IBGE 2004). As species within the genus Ctenomys are primarily known for inhabiting open areas covered mostly by grassy vegetation, sampling individuals in Mato Grosso is interesting because the southernmost parts of Amazon – roughly, western Rondônia and northern/central Mato Grosso – present vegetation characteristic of both the “evergreen forests” of the Amazon to the north and the Cerrado (Savanna), dominated by smaller trees and bush-like vegetation, to the south, the same region where our sampling sites are located. The convergence of the two biomes forms a zone of ecological tension (ZOT) that spans hundreds of kilometers at the southern border of the Amazon (Ackerly et al. 1989; Marimon et al. 2013). This ZOT is likely the product of continuous alternation of temperature and precipitation throughout the Quaternary and is dominated by vegetation characteristic of the Amazon during wet events and by vegetation characteristic of the Cerrado during dry events (Marimon et al. 2013 and references therein). Such dynamics may affect the dispersion of Ctenomys throughout midwest and northern Brazil, if the changes in vegetation composition represent geographical barriers. In addition, some sampling sites were located near the type locality for the species C. nattereri (Cáceres – MT; Miranda Ribeiro, 1914) and C. bicolor (Pimenta Bueno – RO, Stolz et al. 2013).
Therefore, these new sampling sites provide an opportunity to clarify the taxonomic status of poorly known species of Ctenomys located at the northern end of the distribution of the genus, making way for future population genetics and ecological studies. Herein, we build on previous phylogenetic studies and characterize the known localities of Ctenomys from midwest Brazil based on cyt b sequences and geometric morphometrics of the skull, placing the haplotypes representative of the localities sampled into a phylogenetic framework and defining, when possible, taxonomic units.
Material and Methods
Sample Collection and DNA Extraction from Field Specimens
We discovered and sampled ten localities for the genus Ctenomys between the years 2007 and 2017, totaling 92 specimens (Table 1). We captured the specimens using Oneida Victor n° 0 Snap Traps (Zenuto and Busch 1998) under a license from the Brazilian government (IBAMA, Authorization No. 14690–1). We conducted at least one field trip per year, with a minimum sampling effort of 14 days per field trip. All procedures involving the captured animals were allowed by IBAMA (Brazil’s environmental agency) and followed the guidelines of the American Society of Mammalogists (Sikes et al. 2016). Liver samples were collected from 92 individuals distributed among 10 sampling sites in the states of Mato Grosso (MT) and Rondônia (RO) (individuals per locality and geographic coordinates of the sampling sites are detailed in Table 1). Tissue samples were housed at the Laboratory of Cytogenetics and Evolution in the Department of Genetics at Universidade Federal do Rio Grande do Sul (UFRGS) and stored in 96° GL alcohol at −20 °C. We extracted DNA from the samples following the CTAB DNA extraction protocol (Doyle and Doyle 1987), with modifications. All DNA extractions were stored at −20 °C.
Mitochondrial DNA Primers
We amplified partial coding sequences – CDS (1087 bp) –of the cyt b gene (complete CDS: 1146 bp) from 92 individuals using the primer pairs MVZ05 and Tuco06 (Smith and Patton 1999) and Tuco07 and Tuco14 (Wlasiuk et al. 2003). Only forward primers were used for sequencing PCR products.
Cytochrome b PCR conditions and sequencing
We conducted PCR using 100 ng of purified genomic DNA, 0.4 μL of 10 mM forward and reverse primers, 0.4 μL of 10 mM deoxynucleotide triphosphates, 2.0 μL of 10X PCR buffer, 1.6 μL of 50 mM MgCl2 polymerase cofactor, and 0.2 μL of 5 U/uL DNA Taq polymerase (Ludwig Biotec), totaling 20 μL of reaction per sample. PCR cycling conditions were as follows: initial denaturation at 94 °C for 1 minute, followed by 30 cycles of denaturation at 93 °C for 1 minute, primer annealing at 45 °C for 1 minute, and fragment extension at 72 °C for 1.5 min, ending with a final extension at 72 °C for 10 min. To confirm amplification, the PCR products were visualized in 1.5% agarose gel prior to sequencing. Sequencing was conducted abroad (Macrogen Inc., Seoul, Korea).
Mitochondrial DNA Data Sets
We created two cyt b data sets. The global data set comprises all unique haplotypes representative of midwest and northern Brazil, 69 sequences representative of the genus Ctenomys, and two Octodontidae haplotypes downloaded from GenBank (accession numbers: Table S1) used as outgroups; we used this data set to reconstruct a complete phylogeny for Ctenomys, placing Brazilian haplotypes into species groups, and to estimate the time of origin of the most recent common ancestor (MRCA) between taxa of interest. The local data set comprises all haplotypes representative of the sampled localities, ten sequences representative of the boliviensis species group (sensu Parada et al. 2011), and six sequences representative of the species Ctenomys andersoni Gardner et al., 2014, Ctenomys erikacuellarae Gardner et al., 2014, and Ctenomys yatesi Gardner et al., 2014, which form a clade sister to boliviensis (Gardner et al. 2014). We calculated partial phylogenetic reconstructions to assess intragroup phylogenetic relationships and divergence times between taxa belonging to the boliviensis species group; we assumed it was plausible to calculate partial reconstructions for this data set because all haplotypes representative of known sampling sites clustered within the boliviensis species group for all phylogenies constructed using the global data set (see Results). In addition, we also compared estimates obtained through the partial phylogenetic reconstruction with those obtained through the complete phylogenetic reconstruction to assess whether the estimates corroborate each other.
Mitochondrial DNA Analysis
We inspected chromatograms and trimmed sequences using Chromas Lite 2.1.1 (Technelysium Inc.). We aligned the sequences used in both data sets independently through the MUSCLE algorithm (Edgar 2004) implemented in MEGA 6.0 (Tamura et al. 2013) using the default parameters. Best-fit models of molecular evolution for each data set were calculated using JModelTest2 (Guindon and Gascuel 2003; Darriba et al. 2012). The model selected was based on the smallest variation in the logarithm of the Bayesian Information Criteria (BIC). The haplotype (Hd) and nucleotide diversity (Pi) indexes and number of segregating (S) and invariant (I) sites obtained were estimated using DnaSP 5.0 (Librado and Rozas 2009). We used the Kimura-2-parameters (K2P) substitution model with pairwise deletion of gaps and missing data to estimate percentages of sequence divergence between haplotypes for the cyt b gene, with 1000 bootstrap replicates and considering the 1st, 2nd, and 3rd codon positions, in MEGA 6.0. We performed a nonspatial genetic mixture analysis in BAPS 6 (Corander et al. 2006, 2008; Cheng et al. 2011) to estimate the genetic clusters formed by individuals of each locality analyzed with the complete data set for the cyt b gene (92 sequences, 1087 bp), using five iterations and a maximum number of clusters of 20 (K = 20); all other parameters were left as default.
We constructed maximum likelihood (ML) phylogenies (global data set) in MEGA 6.0 using 1000 bootstrap replicates, considering the 1st, 2nd, and 3rd codon positions; all other parameters were left as default. We constructed Bayesian inference (BI)-based phylogenies for both the global and local data sets in BEAST v2.4.0 (Bouckaert et al. 2014) with empirical base frequencies, four gamma categories and data partitioned into the 1st, 2nd, and 3rd codon positions. As the data are not clock-like, a relaxed uncorrelated lognormal clock (Drummond et al. 2006) was used to allow different mutation rates for each branch in the phylogeny while preserving the time-dependent nature of the evolutionary process. A calibrated Yule speciation prior (Heled and Drummond 2011) was implemented to allow calibration of trees calculated with the global data set for both the Ctenomyidae/Octodontidae split (Marshall and Sempere 1993; Vucetich et al. 1999; Verzi 2002; Table 4) and the MRCA for the genus Ctenomys (Verzi et al. 2009; Table 4); we calibrated the local data set with the MRCA estimates for the boliviensis species group obtained through the complete phylogenetic reconstruction (see Table 5). Twenty million iterations in a Markov chain Monte Carlo (MCMC) process were used in each run, sampling a tree every 1000 steps in both the global and local analyses; all other parameters were left as default. Six independent runs were calculated for each data set, and the outputs of the runs were submitted to a 25% burn-in and combined into a single tree file using LogCombiner v2.4.3 (Rambaut and Drummond 2015a). The consensus tree was chosen using TreeAnnotator v.1.8.2 (Rambaut and Drummond 2015b). The log files of all runs were inspected in Tracer v1.6 (Rambaut et al. 2013), to ensure that all parameters had estimated sample size (ESS) values higher than 200.
Skull Geometric Morphometric Analysis
The sample consisted of 67 skulls of adult specimens of Ctenomys from nine sampling sites in midwest and northern Brazil (see Table 1). Skulls from all individuals are housed at the Laboratory of Cytogenetics and Evolution in the Department of Genetics at UFRGS (archive numbers, Table S4). Dorsal, ventral, and lateral left sides of the skulls were photographed with a Canon G10 digital camera at maximum resolution (4416 × 3312) with the macro function activated and without flash. We digitized 29, 30, and 21 morphological landmarks for each individual for the dorsal, ventral and lateral views, respectively (sensu Fernandes et al. 2009), with TPSDig 2.16 (Rohlf 2010). All landmarks plotted were assigned to each individual by the same person (L.E.J.R.). Coordinates were superimposed by the generalized Procrustes analysis (GPA) algorithm (Dryden and Mardia 1998). We symmetrized landmarks in dorsal and ventral views of the skull and used the symmetric component of variation in each view for our analysis (Kent and Mardia 2001; Klingenberg et al. 2002). We used the residuals of GPA-like shape variables as the shape, and we used the centroid size (the square root of the sum of distances of each landmark from the centroid; Bookstein 1991) as a measure of overall skull size. To avoid redundancy of the three views, we summed the centroid sizes of the three views of the skull and used the obtained mean for the analysis.
We tested skull centroid size for normality with Shapiro’s test, sexual dimorphism for size using Student’s t test, and sexual dimorphism for skull shape with multivariate analysis of variance (MANOVA). We tested differences in size among sampling sites with analysis of variance (ANOVA) and used Tukey’s test for pairwise comparisons. We implemented principal component analysis (PCA) to explore shape variation for each view of the skull and multivariate analysis of variance (MANOVA) to test for differences in skull shape between sampling sites. We calculated jackknife cross-validation to generate percentages of correct classification among sampling sites and within hypothesized molecular clades (see Results). We conducted all aforementioned analyses in R software version 3.2.1 (R Core Team 2017) with the geomorph package (Adams and Otárola-Castillo 2013). We used canonical variate analysis (CVA) in MorphoJ (Klingenberg 2011) to generate scatterplots and wireframes to discriminate the individuals and localities sampled in shape space.
Data Availability
All sequences from the localities studied used in the analyses present in this manuscript are available at GenBank (https://www.ncbi.nlm.nih.gov/genbank) under accession numbers MG065617 to MG065634. Detailed individual information on the origin of each sequence we downloaded from GenBank and on the sequences we published is available as supplemental material (Tables S1 and S2, respectively).
Results
Cytochrome b diversity indexes for sampled localities
Partial coding sequences for the cyt b gene (1087 bp) were obtained for 92 individuals. A total of 1083 bp were used in the analysis, excluding 4 bp of missing data. The haplotype diversity (Hd) and nucleotide diversity (Pi) indexes were equal to 0.8830 and 0.01737, respectively. The total number of segregating sites (S) was 69, while 1014 sites were invariant (I). Eighteen individual sequences corresponded to 15 haplotypes representative of the genetic variability of the sampled localities. Out of these 18 sequences, 15 corresponded to exclusive haplotypes, and three were shared among sampling sites (H4: PL and SP; H10: NU1, NU2, and FN; Fig. 1 and Table S2). Therefore, we included 15 sequences representing the total haplotype variability of the sampling sites in the phylogenetic analyses (GenBank accession numbers, Table S2).
Cytochrome b diversity indexes for the global data set
A total of 86 sequences were used in the complete phylogenetic reconstruction analyses with the global data set: 15 haplotype sequences representing the sampling sites of Ctenomys from midwest and northern Brazil, 69 sequences representative of the genus Ctenomys, and two Octodontidae sequences as outgroups. The sequences from GenBank were mostly complete (1140 bp), but some were shorter; a few sequences contained small nucleotide gaps or undetermined nucleotides (‘N’). Overall, 1042 bp were used in the phylogenetic analyses, given that there were 45 sites with missing data. From a total of 84 haplotypes, 577 were variable sites, and 445 were parsimony informative (S) sites. Out of the 1042 sites analyzed, 465 were invariant (I).
Genetic Distances between Sampled Localities from Midwest and Northern Brazil
Genetic distances based on the Kimura-2-parameters model for the partial CDS of the cyt b gene (1087 bp) presented mean values of 0.2% between PL and SP, 0.1% between NU1 and NU2, and 0.7% between NM and TS. The maximum distance reached was 3.8%, between PB and CA. Some subsets of localities had lower sequence divergence between haplotypes within subsets than other subsets of localities: there was a maximum of 1.3% divergence between sequences from the sampling sites PL, CA, and SP, but a maximum of over 2.0% when compared against any other haplotypes. Sequence divergence between the localities NO, NU1, NU2, and FN presented a mean of 0.6 and 1.6% when compared against NM and TS and a mean of over 2.2% when compared to PL, CA, SP, and PB. PB, the only locality known to represent the species C. bicolor (Stolz et al. 2013), diverges from the other localities by a mean of 2.9% (Table 2). Groups of localities, therefore, seem to cluster into clades when the genetic distances between them are considered. We name such clusters by their relative geographic positioning (PL, CA, and SP: Western Clade; NO, NU1, NU2, and FN: Eastern Clade; and NM and TS: Central Clade; Table 3).
Genetic Distances for the Global Data Set
We estimated genetic distances between haplotypes based on the Kimura-2-parameters model (Table 3). Mean genetic distances within clades are lower (Western Clade: 0.7%; Eastern Clade: 0.6%; Central Clade: 0.7%) than those between clades (Western vs. Eastern: 2.7%; Western vs. Central: 2.2%; Eastern vs. Central: 1.6%). The genetic distances between C. bicolor (PB) and all other clades are higher than the distances between clades (Table 3). Genetic distances between the Western Clade and a haplotype representative of C. nattereri from Bolivia (HM777484) present a mean of 1.0%; C. nattereri from Bolivia diverges in over 1.9% of the sequence when compared against the Eastern and Central Clades (C. nattereri vs. Eastern: mean of 2.4%; C. nattereri vs. Central: 2.0%) and in 3.2% of the sequence compared to C. bicolor. Sequences identified as C. boliviensis differ widely from each other (AF007039 and AF007040 compared to AF007037, AF007038, and KJ778554: 5.3%; AF007037 and AF007038 compared to KJ778554: 1.4%) and when compared to C. nattereri, C. bicolor, and the clades specified above (Table 3). However, the haplotypes AF007039 and AF007040 are known to have been misidentified as C. boliviensis, because individuals had a diploid number of 2n = 36, which is distinct from that known from C. boliviensis localities (2n = 42, 44, and 46) and did not cluster with actual C. boliviensis haplotypes in past studies (Lessa and Cook 1998; Mascheretti et al. 2000). Haplotypes of C. goodfellowi differ by a mean of 1.3% from true C. boliviensis (AF007037, AF007038, and KJ778554) but by over 5.0% from the haplotypes AF007039 and AF007040, which is consistent with the findings of Gardner et al. (2014), who synonymized C. goodfellowi with C. boliviensis (Table 3). All other haplotypes representative of Ctenomys diverge in over 6.0% of their sequences compared to the Western, Eastern, and Central Clades; C. nattereri from Bolivia; and C. bicolor (PB) (Table 3).
Nonspatial Genetic Mixture Analysis
The Bayesian inference of hierarchical clustering of sampling sites estimated in BAPS 6 (Corander et al. 2006, 2008; Cheng et al. 2011) yielded six genetic clusters (K = 6), which resemble the clades inferred by genetic distances between sampling sites (PL and SP; CA; NO; NU1, NU2 and FN; PB; and NM and TS; Fig. 2).

Non-spatial genetic mixture analysis implemented in BAPS 6. Localities sampled in this study structure into six genetic clusters (K = 6). Colors from white to dark grey indicate each genetic genetic cluster. From left to right: PL/SP; CA; NO; NU1 NU2, FN; PB; NM, TS
Cytochrome b phylogenies: global data set
The best-fit model of molecular evolution calculated in JModelTest2 for the global data set was Hasegawa-Kishino-Yano (Hasegawa et al. 1985) + gamma + invariant sites (HKY + G + I). Most species groups were recovered with moderate to strong statistical support in both phylogenies, though the estimated times of divergence between most species groups had only moderate to low support (Figs. 3 and 4). All haplotypes from midwest and northern Brazil are included in the boliviensis species group, which is monophyletic, with strong statistical support (Figs. 3a and 4a). Haplotypes from the Western Clade and C. nattereri from Bolivia (HM777484) share an MRCA with moderate support in the ML phylogeny (Fig. 3) and strong support in the BI phylogeny (Fig. 4). Haplotypes from the Eastern Clade share an MRCA with C. bicolor, while haplotypes from the Central Clade are sister taxa to the Eastern Clade plus C. bicolor, with strong statistical support in both phylogenies. All haplotypes from midwest and northern Brazil; the two misidentified haplotypes representative of the species C. boliviensis (AF007039 and AF007040) and one haplotype representative of the species C. nattereri from Bolivia (HM777484) share an MRCA with five C. boliviensis haplotypes (AF007037, AF007038, AF007050, AF007051, and KJ778554) with strong statistical support in both trees. It is important to note that the haplotypes AF007050 and AF007051 are identified in data bases as C. goodfellowi, but since Gardner et al. (2014), this species is considered synonymous with C. boliviensis. Mean estimates for time of divergence and their 95% highest probability densities (HPD95%) between species groups, between the Ctenomyidae and Octodontidae split, and for the MRCA for the genus Ctenomys are summarized in Table 4. The MRCA for the genus Ctenomys was estimated at approximately 3.71 mya; the frater species group is estimated to be the first species group to have originated, approximately 2.18 mya, while the other species groups are more derived. Conversely, talarum and mendocinus are the most recently originated species groups (mean time estimates of 0.52 and 0.64 mya, respectively; Table 4; Figs. 3 and 4).

Maximum Likelihood phylogeny for the Cytochrome b gene (1087 bp) calculated for the global data set. Haplotypes sampled in this study cluster with the boliviensis group of species and share a common ancestor with the Bolivian species Ctenomys boliviensis. Western Clade: PL, CA, SP; Eastern Clade: NO, NU1, NU2, FN; Central Clade: NM, TS; C. bicolor: PB. Species groups: (a)boliviensis; (b) opimus; (c) mendocinus; (d) talarum; (e) torquatus; (f) tucumanus; (g) magellanicus; (h) frater. Bootstrap values under 50 are ommited from the figure

Bayesian Inference based phylogeny for the Cytochrome b gene (1087 bp) for the global data set. Haplotypes sampled for the studied localities cluster within the boliviensis group of species and share a most recent common ancestor (MRCA) with the species Ctenomys boliviensis. Western clade: PL, CA and SP; Eastern Clade: NO, NU1, NU2 and FN; Central Clade: NM and TS; Ctenomys bicolor: PB. Species groups: (a) boliviensis; (b) opimus; (c) mendocinus; (d) talarum; (e) torquatus; (f) tucumanus; (g) magellanicus; (h) frater. Values of posterior probabilities lower than 0.5 were omitted from the figure
There is one incongruence between the topologies of the BI and ML phylogenies related to the C. boliviensis haplotypes AF007039 and AF007040. When comparing these haplotypes to the others attributed to C. boliviensis, the haplotypes AF007039 and AF007040 share an MRCA with C. nattereri and the Western Clade but not with the other haplotypes of C. bicolor. In both cases, however, there is low statistical support for such positioning (Figs. 3 and 4). Statistical support for the topologies including C. steinbachi, C. erikacuellarae, C. andersoni, and C. yatesi as species either within or outside the boliviensis species group is equal to one in the BI phylogeny, while the bootstrap values for such clusters are low (57) and moderate (82) in the ML phylogeny for the more inclusive and exclusive groups, respectively (Figs. 3 and 4). Given the maximum support was obtained when including the species in a monophyletic species group (e.g., boliviensis) in the BI phylogeny, we chose to consider boliviensis as a more inclusive group (species group A; Figs. 3 and 4) as a means to estimate the times of origin for the MRCAs between the taxa of interest while not constraining the analyses to a smaller data set. Therefore, the mean time of divergence for the boliviensis species group was estimated at approximately 1.58 mya (Table 4).
Almost all clades within the boliviensis species group estimated through the complete phylogenetic reconstruction present moderate (0.70–0.89) to high (0.90–1) statistical support for the given best tree topology, except when considering the clustering of individuals within sampling sites and the positioning of C. steinbachi within the species group (posterior probability values less than 0.50). The localities from midwest and northern Brazil share an MRCA with C. boliviensis at approximately 1.17 mya. The MRCA between the localities was estimated to have originated 0.57 mya on average, while the clades were estimated to be somewhat more recent (C. nattereri and Western Clade, 0.50 mya; Eastern Clade, 0.17 mya; Central Clade, 0.20 mya) (Local group, Table 4).
Cytochrome b phylogenies: local data set
Bayesian phylogenies for the local data set were calculated in BEAST v2.4.2 using the HKY + G model of nucleotide substitution with four gamma categories and empirical base frequencies. The mean estimated time of divergence for the boliviensis species group was 1.57 mya. Ctenomys bicolor, C. nattereri, and the Eastern, Western, and Central Clades share an MRCA with C. boliviensis, with a mean estimated time of origin of approximately 1.06 mya. The Eastern and Central Clades originated approximately 0.15 and 0.17 mya, respectively, while the Western Clade plus C. nattereri from Bolivia (HM777484) is estimated to be somewhat older on average (0.24 mya). The mean estimated times for common ancestors between the taxa in the local data set are summarized in Table 5.
The tree topology is also maintained in the phylogeny calculated with the local data set, with the exception of the positioning of C. steinbachi, which clusters as a sister taxon to C. andersoni, C. erikacuellarae, and C. yatesi, albeit with low statistical support (Fig. 5). Almost all nodes representative of valid species and lineages have strong statistical support (posterior probabilities ranging from 0.9 to 1), with the exception of the nodes that separate the species C. steinbachi from C. andersoni, C. erikacuellarae, and C. yatesi (0.45), the Western Clade and C. nattereri from Bolivia from the two haplotypes misidentified as C. boliviensis (AF007039 and AF007040; 0.56) and C. bicolor from the Eastern Clade (0.79).

Bayesian Inference based phylogeny for the Cytochrome b gene (1087 bp) for the local data set. Clades of localities are indicated at the bars to the right. The boliviensis species group is monophyletic with strong statistical support. Posterior probability values lower than 0.5 are ommited from the figure
Geometric morphometric analyses – Shapiro-Wilk’s test indicated that the centroid sizes do not differ between localities (w = 0.97302, P = 0.153) for any of the three views (dorsal: P = 0.153; ventral: P = 0.07; lateral: P = 0.233), except for locality SP, the individuals of which are smaller on average than the ones in other localities (F8; 58 = 6.489; P < 0.001) (Fig. S1). We did not find significant sexual dimorphism for size or shape within or between localities (t = 0.31798, df = 32.362, P = 0.7525; Fig. S1). Therefore, the males and females were pooled together for further analyses.
The PCAs did not show structuring between localities for skull shape (data not shown). Cross-validation tests assigned on average 86%, 68%, and 67% of the individuals to their localities of origin (Table 6) and 91%, 83%, and 64% of the individuals to their respective molecular clade for the dorsal, ventral, and lateral views, respectively (Table 7). There are significant differences among localities for skull shape for the dorsal (Wilk’s λ < 0.0001; F8, 58 = 5.8889; P < 0.001), lateral (Wilk’s λ < 0.0001; F8, 58 = 3.7815; P < 0.001), and ventral (Wilk’s λ < 0.0001; F8, 58 = 3.3744; P < 0.001) views. Pairwise MANOVA revealed significant differences in skull shape between the majority of comparisons for all views (Table S4). The scatterplot of the CVA for the three views of the skull showed different degrees of discrimination for the localities sampled (Fig. S2): in the dorsal view, the localities PB and CA overlap, with the highest negative values for CV1, with the skulls showing a proportionately more-elongated rostrum in positive scores (Fig. S2a). In contrast, NO presents the highest positive values for this axis. All other localities (PL, SP, NU1, NU2, FN, and NM) are differentiated from CA, PB, and NO, clustering with mostly positive values for CV2, though NM presents slightly negative values for CV1; locality FN is differentiated from all other localities by CV2, presenting the highest positive values for that axis. In the ventral view, the localities PL, CA, and SP are differentiated from all other localities by CV1, where sampling sites with positive scores show skulls enlarged in the tympanic bullae (Fig. S2b). NM and NO present values closest to the mean skull shape for the ventral view, e.g., close to zero, while the NU1, NU2, and FN localities are differentiated by CV2. PB is separated from all other localities by CV1, presenting the most negative values for that CV. In the lateral view, the locality NU1 presents the shape closest to the mean shape, while all other sampling sites are separated from each other and from NU1, with values distant from zero for both CVs. PL, FN, and PB are closely distributed in shape-space, presenting the most negative values for CV1 and little variation in skull shape (Fig. S2c).
Discussion
Diversity indices and general patterns of genetic structuring in Ctenomys from midwest and northern Brazil
Two haplotypes are shared between localities within clades (PL and SP: H4; NU1, NU2, and FN: H10), which were in turn proposed based on the genetic distances between haplotypes. In every case, the pairwise genetic distances between haplotypes in a clade were lower than those in any other comparison between haplotypes. Indeed, the clades specified are monophyletic with strong statistical support in all phylogenetic analyses and cluster within the boliviensis species group (group A; Figs. 3, 4 and 5). In addition, the Bayesian mixture analysis structures localities into six genetic groups, consistent with the groups formed when considering the observed shared haplotypes, genetic distances, and tree topologies (Fig. 2). The clustering of the haplotypes from midwest and northern Brazil within the boliviensis group of species was expected, given that C. bicolor (Stolz et al. 2013) and C. nattereri from Bolivia (Parada et al. 2011; Gardner et al. 2014) are known to cluster within this species group.
The mean genetic distances observed between C. nattereri from Bolivia and the Western Clade (1.0%) are similar to the ones observed within the Western Clade localities (0.7%) and within the Eastern Clade localities (0.6%), which we consider intraclade values, as opposed to the values observed between the Eastern and Western Clades (mean of 2.8%), which can be two to three times larger. In contrast, C. nattereri and the Eastern Clade present a mean sequence divergence of 1.9%, while C. bicolor diverges in over 2.2% of the sequence when compared to the Western (mean of 3.5%), Eastern (mean of 2.5%), and Central (2.3%) Clades. Such genetic divergence between the haplotypes sampled, combined with the evidence of shared haplotypes, which is reflected by the structuring among haplotypes of the sampled localities, and with the phylogenetic structuring of clades into monophyletic groups with strong statistical support indicate that the clades are representative of unique lineages of Ctenomys in midwest Brazil. However, proposing thresholds for sequence divergence to delimitate intra- and interspecific genetic distances is difficult, as the same marker may evolve at different rates in different taxa (Avise 1994). Few studies have estimated sequence divergence thresholds for mitochondrial genes. Tobe et al. (2010) compared cytochrome oxidase I (COI) and cytochrome b (cyt b) sequences for 217 mammalian taxa and concluded that at the specific level, cyt b estimates generally correctly assign individuals to their species better than do COI estimates, especially when considering 1.5–2.5% sequence divergence as the threshold to distinguish taxa. Such estimates would imply that our proposed clades present genetic distances within them below the 1.5–2.5% threshold, while the distances between clades can reach values that surpass this threshold, meaning that the clades may represent different species or, at least, represent lineages in the process of speciation. Thus, the distances between C. bicolor and the clades are either near or above 2.5%, which is expected because C. bicolor is considered a valid species (see Table 3; Figs. 3, 4 and 5).
Phylogenetic trees, skull geometric morphometrics, and diversity of Ctenomys in midwest and northern Brazil
The phylogenies calculated using BI and ML methods yield very similar topologies and recover all eight species groups proposed by Parada et al. (2011) with moderate to strong statistical support (Figs. 3, 4 and 5). There are minor incongruences between the phylogenies estimated with the two methods, but they are restricted to the species positioning within species groups, and none of them directly relate to our taxa of interest. Our analyses show that the boliviensis species group is one of the oldest species groups to have originated (mean estimate of 1.57 mya; Table 5), considering the estimate generated through the local phylogeny. The lack of resolution for the basal nodes on the phylogenies (bootstrap values under 50 and posterior probabilities under 0.5) demonstrates the rapid radiation of Ctenomys throughout South America, as has been pointed out by other authors (Lessa and Cook 1998; Castillo et al. 2005; Parada et al. 2011). The estimates of origin of MRCAs for the boliviensis species group obtained from both the complete and partial phylogenetic reconstructions indicate that the mean and highest probability density (HPD95%) values obtained are consistent between analyses, with minor differences. This demonstrates that the results are reproducible and that the parameters used to reconstruct phylogenies, most notably the calibration points and the enforcing of monophyly of the genus Ctenomys, were adequate.
The CVAs for dorsal and ventral views of the skull more closely differentiate the localities in a phylogeny-like pattern: the CV1 for the ventral view of the skull segregates the sampling sites into groups resembling the molecular clades, while in the dorsal view, CV2 also discriminates the sampling sites in a geographical pattern (Fig. S2a and S2b). The CVA for the lateral view of the skull differentiates the localities into single-locality groups, with low variance for skull shape within them (Fig. S2c). The percentages of individuals correctly assigned to their localities of origin through jackknife cross-validation analyses suggest that differences in the dorsal view of the skull best differentiate the sampling sites from one another (86%), while the ventral and lateral views correctly differentiate the sampling sites less often (68 and 67%, respectively; Table 6). However, testing the assignment of individuals based on the proposed phylogenetic clades increased the frequency of correct classification for both the dorsal (91%) and ventral (83%) views, suggesting that skull shape is influenced partially by demography (Table 7), because the increase in correct classification reflects the common origin of the individuals of the localities within a clade. Patterns of discrimination between sampling sites through the CVA analysis corroborate the results obtained through the MANOVA analysis, in which there are significant differences in skull shape for most pairwise comparisons in all three views analyzed (Table S4).
Therefore, the geometric morphometric analysis of the skulls of individuals from our sampling sites demonstrates that variation in skull shape may contain both environmental and phylogenetic signals. Because mitochondrial genes are considered to evolve under near neutrality, and thus are widely used as molecular markers for estimating evolutionary processes dependent on geographical barriers, gene flow and migration (e.g., demographic effects), environmental pressures selecting for a given cranial structure may not reflect a pattern of cranial structure in which variation is caused by demographic effects but rather a pattern that reflects adaption or both adaption and demography. Indeed, the structure inferred through the morphometric analysis for both the ventral and dorsal views of the skull closely resembles the structure found in the phylogenetic analysis, while the morphometric analysis with the lateral view of the skull does not, indicating the influence of selection on skull shape.
There are many possible explanations for the differences in skull shape that do not match the patterns of structure inferred from the phylogenies as well as for the lack of differences between individuals in localities that do belong to different phylogenetic clades; for example, convergence in the feeding apparatus in rodents inhabiting similar habitats is common (Samuels 2009). Therefore, if the habitats present similar ecological pressures, then the differences in skull shape between individuals of localities representative of distinct phylogenetic lineages can be more influenced by environmental pressures than by demography alone. Environmental pressures, such as the hardness of soil, can select for skull shape in fossorial rodents (Marcy et al. 2016), which requires a derived skull morphology with procumbent incisors for digging (Stein 2000); indeed, the bulk density of soils is correlated with bite force in Ctenomys, with species that inhabit denser soils generally exhibiting higher bite force measurements (Borges et al. 2017). Alternatively, allometry effects can influence intraspecific skull shape in conjunction with environmental effects, such as primary production and precipitation, or demographic effects, including restricted gene flow and reduced population size (Maestri et al. 2016). Conversely, the times of origin for the Brazilian clades appear to be recent (Table 5), in which case it is possible that not enough time has passed for the lineages to accumulate differences in skull shape. This demographic hypothesis could explain the subtle structural differences found between the three views of the skull. Alternatively, environmental pressures may be selecting for skull shape.
Overall, patterns of phylogenetic structure, sequence divergence between haplotypes, and skull shape differences between individuals within the sampled localities reveal two major lineages in midwest Brazil: i) a lineage including the haplotypes from the Western Clade plus C. nattereri from Bolivia (HM777484), and ii) a lineage including all haplotypes from the Central and Eastern Clades and C. bicolor. Given that C. bicolor is a valid species and is positioned intermediately relative to the Eastern and Central Clades in all phylogenetic analyses, both clades can be considered independent lineages. As the Eastern Clade sampling sites NU1, NU2, and FN are located near Xingu National Park, one of the largest and most conserved protected areas in Brazil, and the Central Clade sampling sites are central to the known distribution of Ctenomys in midwest and northern Brazil, we name the lineages C. sp. “xingu” and C. sp. “central,” respectively.
Potential Shortcomings of the Analyses and Future Directions
Mitochondrial genes have been used extensively in phylogenetic studies due to their mode of inheritance, lack of recombination, and reduced effective sample size, which facilitate coalescence in a phylogeny (Neigel and Avise 1987; Avise 2000). In the case of Ctenomys, mitochondrial genes have been instrumental for defining species and confirming the monophyly of the genus. However, time estimates using mitochondrial markers should be interpreted with caution, because these markers may have broad confidence intervals that may limit inference of the ages of phylogenetic clades (Ballard and Whitlock, 2004 and references therein). In any case, phylogenetic approaches in Ctenomys using the cytochrome b gene have yielded more reliable estimates of the origin of both the genus and its species groups than the nuclear markers that have been tested, based on the estimated age of the oldest known fossil (Lessa and Cook 1998; Castillo et al. 2005; Parada et al. 2011).
Ctenomys presents high levels of karyotype heterogeneity, with diploid numbers ranging from 2n = 10 in C. steinbachi to 2n = 70 in Ctenomys dorbignyi Contreras and Contreras, 1984 (Anderson et al. 1987; Woods and Kilpatrick 2005), although the species generally have species-specific karyotypes (Reig et al. 1990, but see Lopes et al. 2013). Therefore, knowing how localities structure karyotype variability is essential to corroborate molecular and morphological data to define and describe species within the genus Ctenomys. We concur with Parada et al. (2011) and Freitas (2005) in that additional nuclear markers, such as the ones used by Castillo et al. (2005) and Upham and Patterson (2012), should be sequenced genus-wide to corroborate the karyotype, morphological, and mitochondrial DNA data. Additionally, sequencing additional genes can better estimate species trees, thus mitigating possible incongruences between species and gene trees, and can provide more reliable evidence for dating times of divergence between lineages with narrower confidence intervals.
Conclusions
Molecular phylogenetics and geometric morphometrics approaches have allowed us to resolve phylogenetic lineages and corroborate the presence of C. bicolor and C. nattereri in midwest and northern Brazil. We identify two independent lineages: C. sp. “xingu” and C. sp. “central.” At the moment, a lack of karyotype data does not allow us to resolve lineages into defined species; therefore, sampling additional localities of Ctenomys from midwest and northern Brazil, and thus increasing the sample size for molecular and morphometric analysis and allowing for the karyotyping of individuals is essential to accomplish this task. In addition to increasing sample size, future population genetics and ecological studies involving Ctenomys in Brazil will require more molecular markers, both those that are neutral and those under selection, to estimate population parameters, understand genetic mechanisms of local adaption, and define evolutionary significant units sensu Moritz (1994). For example, next-generation sequencing (NGS) methods coupled with restriction enzyme digestion (Davey et al. 2011 and references therein), such as RADseq (Baird et al. 2008) and its derivatives (ddRADseq, Peterson et al. 2012), could be employed to generate sets of hundreds to thousands of neutral and potentially adaptive loci, allowing for the fine-tuning of phylogenies (Cariou et al. 2013; Emerson et al. 2010) and identification of polymorphisms that could account for the genetic basis of local adaption (Manthey and Moyle 2015).
References
Ackerly DD, Thomas WMW, Ferreira CAC, Pirani JR (1989) The forest – cerrado transition zone in southern Amazonia: results of the 1985 Projeto Flora Amazonica Expedition to Mato Grosso. Brittonia 41(2): 113–128
Adams DC, Otárola-Castillo E (2013) Geomorph: an r package for the collection and analysis of geometric morphometric shape data. Methods Ecol Evol 4:393–399
Anderson S, Yates TL, Cook JA (1987) Notes on Bolivian mammals, 4: the genus Ctenomys (Rodentia: Ctenomyidae) in the eastern lowlands. Am Mus Novitates 2891:1–20
Avise JC (1994) Molecular Markers, Natural History, and Evolution. Chapman & Hall, New York
Avise JC (2000) Phylogeography: The History and Formation of Species. Harvard University Press, Cambridge
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, Selker EU, Cresko WA, Johnson EA (2008) Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One 3(10) e3776. https://doi.org/10.1371/journal.pone.0003376
Ballard JWO, Whitlock MC (2004) The incomplete natural history of mitochondria. Mol Ecol 13: 729–744. https://doi.org/10.1046/j.1365-294X.2003.02063.x
Bidau CJ, Ávila-Pires FD (2009) On the type locality of Ctenomys bicolor Miranda Ribeiro, 1914 (Rodentia: Ctenomyidae). Mastozool neotrop 16:445–447.
Bidau CJ (2015) Family Ctenomyidae. In: Patton, JL, Pardiñas, UFJ, D’Elía G (eds) Mammals of South America Vol. 2: Rodents. University of Chicago Press, Chicago
Bookstein FL (1991) Morphometric Tools for Landmark Data: Geometry and Biology. Cambridge University Press, London
Borges LR, Maestri R, Kubiak BB, Galiano D, Fornel R, Freitas TRO (2017) The role of soil features in shaping the bite force and related skull and mandible morphology in the subterranean rodents of genus Ctenomys (Hystricognathi: Ctenomyidae). J Zool 301:108–117. https://doi.org/10.1111/jzo.12398
Bouckaert R, Heled J, Kühnert D, Vaughan T, Wu CH, Xie D, Suchard MA, Rambaut A, Drummond AJ (2014) BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol 10(4): e1003537. https://doi.org/10.1371/jornal.pcbi.1003537
Cariou M, Duret L, Charlat S (2013) Is RAD-seq suitable for phylogenetic inference? An in silico assessment and optimization. Ecol Evol 3(4): 846–852. https://doi.org/10.1002/ece3.512
Castillo AH, Cortinas MN, Lessa EP (2005) Rapid diversification of South American tuco-tucos (Ctenomys; Rodentia, Ctenomyidae): contrasting mitochondrial and nuclear intron suquences. J Mammal 86:170–179
Cheng L, Connor TR, Aanensen, Spratt BG, Corander J (2011) Bayesian semi-supervised classification of bacterial samples using MLST databases. BMC Bioinformatics 12:302
Corander J, Marttinen P, Mäntyniemi S (2006) Bayesian identification of stock mixtures from molecular marker data. Fishery Bull 104: 550–558
Corander J, Marttinen P, Sirén J, Tang J (2008) Enhanced Bayesian modelling in BAPS software for learning genetic structures of populations. BMC Bioinformatics 9:539
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nature Methods 9(8): 772
Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, Blaxter ML (2011) Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nature Reviews Genetics 12(7):499–510. https://doi.org/10.1038/nrg3012
D’Elía G, Lessa EP, Cook JA (1999) Molecular phylogeny of tuco-tucos, genus Ctenomys (Rodentia, Octodontidae): evaluation of the mendocinus species group and the evolution of asymmetric sperm. J Mammal Evol 6: 19–38
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19:11–15
Dryden IL, Mardia KV (1998) Statistical Shape Analysis. John Wiley & Sons, New York
Drummond AJ, Ho SYW, Phillips MJ, Rambaut A (2006) Relaxed phylogenetics and Dating with Confidence. PLoS Biol 4:e88
Edgar R (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Emerson KJ, Merz CR, Catchen JM, Hohenlohe PA, Cresko WA, Bradshaw WE, Holzapfel CM (2010) Resolving postglacial phylogeography using high-throughput sequencing. Proc Natl Acad Sci USA 107 (37):16196–16200. https://doi.org/10.1073/pnas.1006538107
Fernandes FA, Fornel R, Cordeiro-Estrela P, Freitas TRO (2009) Intra- and interespecific skull variation in two sister species of the subterranean rodent genus Ctenomys (Rodentia, Ctenomyidae): coupling geometric morphometrics and chromosomal polymorphism. Zool J Linn Soc 155: 220–237
Freitas TRO (2001) Tuco-tucos (Rodentia, Octodontidae) in southern Brazil: Ctenomys lami spec. nov. separated from C. minutus Nehring 1887. Stud Neotrop Fauna Environ 36: 1–8
Freitas TRO (2005) Analysis of skull morphology in 15 species of the genus Ctenomys, including seven karyologically distinct forms of Ctenomys minutus (Rodentia: Ctenomyidae). In: Lacey EA, Myers P (eds) Mammalian Diversification: From Chromosomes to Phylogeography (A Celebration of the Career of James L. Patton). Univ Calif Publ Zool 133:131–154
Freitas TRO (2016) Family Ctenomyidae. In: Wilson DE, Lacher TE Jr, Mittermeier RA (eds) Handbook of the Mammals of the World: Lagomorphs and Rodents I, Vol. 6. Lynx Editions, Barcelona, pp 499–534
Freitas TRO, Fernandes FA, Fornel R, Roratto PA (2012) An endemic new species of tuco-tuco, genus Ctenomys (Rodentia: Ctenomyidae), with a restricted geographic distribution in southern Brazil. J Mammal 93:1355–1367
Gardner SL, Salazar-Bravo J, Cook JA (2014) New species of Ctenomys (Rodentia: Ctenomyidae) from the lowlands and central valleys of Bolivia. Spec Publ Mus Texas Tech Univ 62:1–34
Guindon S, Gascuel O (2003) A simple, fast and accurate method to estimate large phylogenies by maximum-likelihood. Syst Biol 52:696–704
Hasegawa M, Kishino H, Yano T (1985) Dating the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol 22:160–174
Heled J, Drummond AJ (2011) Calibrated tree priors for relaxed phylogenetics and divergence time estimation. Syst Biol 61:138–149
IBGE (2004) Instituto Brasileiro de Geografia e Estatística. Maps of Biomes and Vegetation. Available at: ftp://ftp.ibge.gov.br/Cartas_e_Mapas/Mapas_Murais/
Kent JT, Mardia KV (2001) Shape, Procrustes tangent projections and bilateral symmetry. Biometrika 88 (2):469–485. https://doi.org/10.1093/biomet/88.2.469
Klingenberg CP (2011) MorphoJ: an integrated software package for geometric morphometrics. Mol Ecol Resources 11: 353–357
Klingenberg CP, Barluenga M, Meyer A (2002) Shape analysis of symmetric structures: quantifying variation among individuals and asymmetry. Evolution 56:1909–1920
Lacey EA, Patton JL, Cameron GN (2000) Life Underground: The Biology of Subterranean Rodents. University of Chicago Press, Chicago and London, 449 pp
Lessa EP, Cook JA (1998) The molecular phylogenetics of tuco-tucos (genus Ctenomys, Rodentia: Octodontidae) suggests an early burst of speciation. Mol Phylogenet Evol 9 (1):88–99
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Lichtenstein H (1830) Darstellung neuer oder wenig bekannter. Saugethiere in Abbildung und Beschriebungen. Luderitz edit, Berlin (1827–31)
Lopes CM, Ximenes SSF, Gava A, Freitas TRO (2013) The role of chromosomal rearrangements and geographical barriers in the divergence of lineages in a South American subterranean rodent (Rodentia: Ctenomyidae: Ctenomys minutus). Heredity 111:293–305. https://doi.org/10.1038/hdy.2013.49
Maestri R, Fornel R, Gonçalves GL, Geise L, Freitas TRO, Carnaval AC (2016) Predictors of intraspecific morphological variability in a tropical hotspot: comparing the influence of random and non-random factors. J Biogeogr 43:2160–2172
Manthey JD, Moyle RG (2015) Isolation by environment in White-breasted Nuthatches (Sitta carolinensis) of the Madrean Archipelago sky islands: a landscape genomics approach. Mol Ecol 24 (14): 3628–3638. https://doi.org/10.1111/mec.13258
Marcy AE, Hadly EA, Sherratt E, Garland K, Weisbecker V (2016) Getting a head in hard soils: convergent skull evolution and divergent allometric patterns explain shape variation in a highly diverse genus of pocket gophers (Thomomys). BMC Evol Biol 16:207. https://doi.org/10.1186/s12862-016-0782-1
Marimon BS, Marimon-Junior BH, Feldpausch TR, Oliveira-Santos C, Mews HA, Lopez-Gonzalez G, Lloyd J, Franczak DD, Oliveira EA, Maracahipes L, Miguel A, Lenza E, Phillips OL (2013) Disequilibrium and hyperdynamic tree turnover at the forest–cerrado transition zone in southern Amazonia. Plant Ecol Diversity 7(1–2):281–292. https://doi.org/10.1080/17550874.2013.818072.
Marshall LG, Sempere T (1993) Evolution of the Neotropical Cenozoic land mammal fauna in its geochronologic, stratigraphic, and tectonic context. In: Goldblatt P (ed) Biological Relationships Between Africa and South America. Yale University Press, New Haven, pp 329–392
Mascheretti S, Mirol P, Gimenez M, Bidau C, Contreras J, Searle J (2000) Phylogenetics of the speciose and chromosomally variable genus Ctenomys (Ctenomyidae, Octodontoidea), based on mitochondrial cytochrome b sequences. Biol J Linn Soc 70:361–376
Miranda-Ribeiro A De (1914) Zoologia. Commisão de Linhas Telegráphicas Estratégicas de MattoGrosso ao Amazonas. Annexo 5, Historia Natural; publ no 17, Mammíferos. 49 pp + Append, 3 pp + 25 pls
Moritz C (1994) Defining ‘evolutionary significant units’ for conservation. Trends Ecol Evol 9 (10):373–375
Neigel JE, Avise JC (1987) Phylogenetic relationships of mitochondrial DNA under various demographic models of speciation. In: Nevo E, Karlin S (eds) Evolutionary Processes and Theory. Academic Press, New York, pp 515–534. https://doi.org/10.1016/B978-0-12-398760-0.50026-2
Nowak RM (1999) Walker’s Mammals of the World. 6th edition, volume 2. Johns Hopkins University Press, Baltimore
Parada A, D’Elía G, Bidau CJ, Lessa EP (2011) Species groups and the evolutionary diversification of tuco-tucos, genus Ctenomys (Rodentia: Ctenomyidae). J Mammal 92(3):671–682
Patton JL, Pardiñas UFJ, D’Elía, G (2015) Mammals of South America, Vol. 2. University of Chicago Press, Chicago
Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE (2012) Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS One 7(5)e37135. doi:https://doi.org/10.1371/journal.pone.0037135
R Core Team (2017) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/
Rambaut A, Drummond AJ (2015a) LogCombiner v1.8.2. http://beast.bio.ed.ac.uk
Rambaut A, Drummond AJ (2015b) TreeAnnotator v1.8.2: MCMC Output analysis. http://beast.bio.ed.ac.uk
Rambaut A, Suchard M A, Xie W, Drummond AJ (2013) Tracer v.1.6.0: MCMC Trace Analysis Tool. http://beast.bio.ed.ac.uk
Reig OA, Busch C, Ortells MO, Contreras JR (1990) An overview of evolution, systematics, population biology, cytogenetics, molecular biology and speciation in Ctenomys. In: Nevo E, Reig OA (eds) Evolution of Subterranean Mammals at the Organismal and Molecular Levels. Wiley- Liss, New York, pp 71–96
Rohlf FJ (2010) TPSDig 2.16. Stony Brook New York: Department of Ecology and Evolution, State University of New York at Stony Brook
Sikes RS, the Animal Care and Use Committee of the American Society of Mammalogists (2016) 2016 Guidelines of the American Society of Mammalogists for the use of wild mammals in research and education. J Mammal 97(3):663–688
Samuels JX (2009) Cranial morphology and dietary habits of rodents. Zool J Linn Soc 156:864–888
Slamovits CH, Cook JA, Lessa EP, Rossi MS (2001) Recurrent amplifications and deletions of satellite DNA accompanied chromosomal diversification in South American tuco-tucos (Genus Ctenomys, Rodentia: Octodontidae): a phylogenetic approach. Mol Biol Evol 18:1708–1719
Smith MF, Patton JL (1999) Phylogenetic relationships and the radiation of sigmodontine rodents in South America: evidence from cytochrome b. J Mammal Evol 6(2): 89–128 S
Stein B (2000) Morphology of subterranean rodents. In: Lacey AP, Patton JL, Cameron GN (eds) Life Underground: The Biology of Subterranean Rodents. University of Chicago Press, Chicago, pp 19–60
Stolz JFB, Gonçalves GL, Leipnitz L, Freitas TRO (2013) DNA-based and geometric morphometric analysis to validate species designation: a case study of the subterranean rodent Ctenomys bicolor. Genet Mol Res 12(4): 5023–5037
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Tobe SS, Kitchener AC, Linacre AMT (2010) Reconstructing mammalian phylogenies: a detailed comparison of the cytochrome b and cytochrome oxidase subunit I mitochondrial genes. PLoS One 5(11): e14156. https://doi.org/10.1371/journal.pone.0014156
Travi VH (1981) Nota prévia sobre nova espécie do gênero Ctenomys Blainville, 1826 (Rodentia: Ctenomyidae). Iheringia 60: 123–124
Upham NS, Patterson BD (2012) Diversification and biogeography of the Neotropical caviomorph lineage Octodontoidea (Rodentia: Hystrichognathi). Mol Phylogenet Evol 60-2: 417–429. https://doi.org/10.1016/j.ympev.2012.01.020
Verzi, DH (2002) Patrones de evolución morfológica en Ctenomyinae (Rodentia, Octodontidae). Mastozool neotrop 9:309–328.
Verzi DH, Olivares AI, Morgan CC (2009) The oldest South American tuco-tuco (late Pliocene, northwestern Argentina) and the boundaries of the genus Ctenomys (Rodentia, Ctenomyidae). Mammal Biol 75:243–252
Vucetich MG, Verzi DH, Hartenberger JL (1999) Review and analysis of the radiation of the South American Hystricognathi (Mammalia, Rodentia). Paleontology 329:763–769
Wagner A (1848) Beiträge zur Kenntniss der Arten von Ctenomys. Archiv für Naturgeschichte 14:72–78
Wlasiuk G, Garza JC, Lessa EP (2003) Genetic and geographic differentiation in the Rio Negro tuco-tuco (Ctenomys rionegrensis): inferring the roles of migration and drift from multiple genetic markers. Evolution 57:913–926
Woods CA, Kilpatrick CW (2005) Infraorder Hystricognathi. In: Wilson DE, Reeder DM (eds) Mammal Species of the World. (3rd ed.). Johns Hopkins University Press, Baltimore, pp 1538–1600
Zenuto RR, Busch C (1998) Population biology of the subterranean rodent Ctenomys australis (tuco-tuco) in a coastal dunefield in Argentina. Z Säugetierk 63:357–367
Acknowledgments
We thank all colleagues from the Laboratory of Cytogenetics and Evolution (LACE) for their invaluable support in reviewing this manuscript. We also thank Professors Andreia Turchetto, Gislene Lopes Gonçalves, and Renan Maestri for reviewing the methods, results, and discussion sections of this paper. L.T.L. and L.E.J.R. received scholarships from the Comissão de Aperfeiçoamento de Pessoal de Nível Técnico e Superior (CAPES), and T.R.O.F. received research grants from CAPES, Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul (FAPERGS).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Leipnitz, L.T., Fornel, R., Ribas, L.E.J. et al. Lineages of Tuco-Tucos (Ctenomyidae: Rodentia) from Midwest and Northern Brazil: Late Irradiations of Subterranean Rodents Towards the Amazon Forest. J Mammal Evol 27, 161–176 (2020). https://doi.org/10.1007/s10914-018-9450-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10914-018-9450-0