
Abstract
Inflammation of the adipose tissues occurs in association with obesity. This inflammatory process leads to the induction of cyclooxygenase-2 (COX-2) expression and a consequent elevation in prostaglandin (PG) production, which, together with proinflammatory cytokines, induce aromatase expression and estrogen synthesis. Infiltrating macrophages support the growth of breast epithelial cells and vascular endothelial cells by producing a milieu of cytokines and growth factors. This scenario creates a microenvironment favorable to breast cancer growth and invasion. The eicosanoids promote further development and growth of breast cancers indirectly by the induction of aromatase, particularly in estrogen positive breast cancers, or by direct stimulatory effect of PGE2 and lipoxygenase (LOX) products on the more aggressive, estrogen-independent tumors. Beyond this, the local production of estrogens and proinflammatory cytokines which occurs in association with breast adipose tissue inflammation, and consequent activation of the estrogen receptor and nuclear factor-κB, provides a mechanism by which breast cancers develop resistance to selective estrogen receptor modulation and aromatase inhibitor therapy. The obesity-inflammation-eicosanoid axis in breast cancer does offer a therapeutic target for the prevention of relapse in breast cancer by improving the efficacy of antiaromatase therapy using COX/LOX inhibitors; however, careful consideration of menopausal status and obesity in patients is warranted.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Epidemiological studies have established that obesity is a risk factor for breast cancer in postmenopausal women, whereas the prevalence of premenopausal breast cancer is either unrelated to obesity, or, in younger women, has an inverse relationship. We will not be re-examining these relationships here, as they have been subjected to extensive review (for example [1–5]).
A major element of the mechanism by which obesity promotes postmenopausal breast cancer development involves extraglandular estrogen synthesis and in consequence the patients frequently have estrogen-dependent, estrogen receptor (ER)-expressing tumors [6]; in premenopausal obese women the tumors that do develop are more likely to be ER-negative [6, 7].
In contrast to disease risk, pre-existing obesity and postoperative weight gain are related to a poor prognosis in breast cancer regardless of menopausal or ER/progesterone receptor (PR) status [8]. Two distinct mechanisms may be involved: in postmenopausal women, elevated estrogen production in the adipose tissue is again most likely responsible, at least in part, for the adverse effect, whereas in premenopausal women this explanation is unlikely because the principal source of estrogens is their high level of secretion by the ovaries. An alternative lies with a number of nonsteroidal hormones and growth factors, such as leptin, insulin, tumor necrosis factor-α (TNF-α) and the eicosanoids, the production of which is elevated in obesity and that have been shown to stimulate breast cancer growth, invasion, and metastasis. A principal objective of our discussion is to explore further these relationships between breast cancer risk and disease outcome, receptor status and tumor biology.
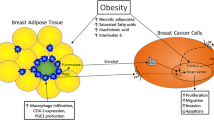
One unifying topic in this review is the role of chronic inflammation in breast cancer causation and biology. Inflammation of the adipose tissues occurs in association with obesity and this, rather than adiposity per se, may provide the mechanisms involved in the causation of a range of chronic diseases, including type 2 diabetes and some cancers. In the breast, this inflammatory process leads to the induction of cyclooxygenase-2 (COX-2) expression and a consequent elevation in prostaglandin (PG) production, which, together with proinflammatory cytokines such as TNF-α, can induce aromatase expression and estrogen synthesis, so creating local conditions favorable to breast cancer growth and invasion [9, 10].
Three families of eicosanoids, PGs, hydroxyeicosatetraenoic acids (HETEs), and leukotrienes (LTs), all products of omega-6 polyunsaturated fatty acid metabolism, have important roles in obesity-associated adipose tissue inflammation [11–13]. In addition to the induction of aromatase and estrogen biosynthesis by PGE2, the eicosanoids have been reported to promote the development and growth of breast cancers directly through their mitotic and anti-apoptotic activities, stimulate tumor cell invasion, and induce tumor-related angiogenesis. Thus, the eicosanoids constitute the third component of our tripartite axis.
Breast Cancer Risk, BMI and Body Fat Distribution
Obesity is usually considered in terms of the body mass index (BMI), which is calculated as body weight (kg)/height (m2). The WHO criteria for classifying adiposity provide four subcategories, <18.5 (underweight); 18.5–24.9 (normal); 25.0–29.9 (overweight); ≥30.0 kg/m2 (obese), according to which 28 % of American women were overweight and 36 % were obese for the years 2009–2010 [14].
The principal sites of body fat accumulation are the upper abdomen and around the hips and thighs. Abdominal adiposity comprises the superficial subcutaneous, deep subcutaneous, and visceral stores, which have different levels of metabolic activity. Visceral fat secretes more cytokines than subcutaneous adipose tissue [15] and the pathological and biochemical changes associated with chronic inflammation are more prominent in visceral than subcutaneous abdominal fat [16]. However, in the present context it should be borne in mind that the modes of action of the estrogens, adipokines and cytokines in breast cancer may all be mediated by their local paracrine and even autocrine activities [17, 18], in which case their production in the breast adipose tissue becomes of prime importance.
In epidemiological studies, body fat distribution has been most often assessed by determining the ratio of the waist-to-hip circumference (WHR) and by this criterion the adverse effect of adiposity on postmenopausal breast cancer risk was shown to apply specifically to women with upper body (“central”) obesity [1, 19, 20]. The data from a large prospective study performed in the United States showed that both WHR and waist circumference were positively associated with risk, but only in postmenopausal women who had never received hormone-replacement therapy [21], suggesting that estrogen administration after the menopause obscures any adverse effect attributable to excessive adipose tissue estrogen production.
Breast Cancer Prognosis and Obesity
Epidemiology
Two recent reviews and meta-analyses have confirmed earlier reports (reviewed in [2, 22]) that there is a positive association between the adiposity and breast cancer prognosis. Protani et al. [23] performed a meta-analysis that included 43 studies of patients diagnosed with breast cancer between 1963 and 2005. It showed that both overall and breast cancer-specific survival were worse in obese than in non-obese women, and that menopausal status had no significant modifying effect. That the relationship applied to breast cancer-specific as well as overall survival is an important point because it eliminates the possibility that it was due to the influence of obesity on the risk of other life-threatening diseases. A later meta-analysis by Niraula et al. [8], which focused on menopausal and hormone receptor status, confirmed that the association of obesity with breast cancer prognosis included a reduction in breast cancer-specific survival and that it applied equally to premenopausal and postmenopausal women. Since this meta-analysis, Kamineni et al. [24] have published a retrospective cohort study of 485 women aged ≥40 years with stage I (75.3 %) or II breast cancer: in 77.8 % of the obese and 61.8 % of normal weight women, the cancers were detected by screening mammography, so avoiding any confounding effect of delayed diagnosis due to obesity. The obese women (BMI ≥ 30.0 kg/m2) were found to be at an increased risk of recurrence and breast cancer-specific death compared to normal weight women (BMI <25.0 kg/m2); there was no association with all-cause mortality in this study. A National Surgical Adjuvant Breast and Bowel Project (NSABP) multicenter study reported by Dignam et al. [25] was included in the meta-analysis by Niraula et al. [8]. Their 4,077 patients, 55 % of whom were premenopausal or perimenopausal, all had lymph node-negative, ER-negative breast cancer and received one of several multidrug adjuvant chemotherapy regimens. The obese group of patients had a shorter disease-free survival, an increased risk of recurrence in the contralateral breast, and higher all-cause, but not breast cancer-specific, mortality. The investigators noted that studies of obesity and breast cancer prognosis in a clinical trial setting, including their own, have consistently shown a weaker effect of obesity on prognosis than those performed outside of large randomized trials, which they suggested was due to better adherence to the full-dose chemotherapeutic regimen in the obese patients.
The meta-analysis by Protani et al. [23] found that a high WHR was also related to reduced breast cancer-specific survival, with no significant influence of menopausal status; a similar association was seen in a prospective study of 586 women by Borugian et al. [26], but here the association was restricted to ER-positive postmenopausal breast cancer. Abrahamson et al. [27] performed an 8 to10 year follow-up study of 1,254 women aged 20–54 years when diagnosed with invasive breast cancer, and found the all-cause mortality to be increased in those who were obese (BMI ≥ 25 kg/m2, when interviewed), with a similar strong association for the highest versus the lowest quartile of current WHR. The data were not provided in the report, but the same association between greater body size and lower survival was observed when the analysis was restricted to breast cancer specific mortality. Also, although the data were available, survival was not examined in relation to the ER status in this study.
Estrogen Receptors, Progesterone Receptors, and HER-2
The expression of ER and PR by breast cancer epithelial cells not only provides biomarkers of estrogen dependence and likely responsiveness to antiestrogen therapy, but is also predictive of a good prognosis [28]. Breast cancers that are positive for both receptors respond better than ER-positive/PR-negative tumors to selective ER modulator treatment, which in the past was ascribed to the need for a functioning ER for PR synthesis, so that an ER-positive/PR-negative phenotype was indicative of a non-functioning ER. More recently, it has been recognized that this combination may be the result of downregulation of the PR by growth factors such as insulin-like growth factor-I and epidermal growth factor [29], and occurs particularly with tumors that also show amplification of epidermal growth factor receptor-2 (HER-2) or epidermal growth factor receptor-1 (EGFR) [30].
Tumors that express both ER and PR comprise approximately 50 % to 60 % of all breast cancers from European and non-Hispanic white American patients, but the prevalence is significantly less in tumors from African-American, Hispanic-black, and Hispanic white women [31]. The incidence of ER/PR-positive breast cancer is higher after the menopause, but it also increases with age within each menopausal category [32]. The presence of obesity is associated with an increased incidence of ER/PR-positive breast cancer in postmenopausal women [6, 33–35], consistent with these tumors developing in the presence of estrogens synthesized by high levels of aromatase activity in the adipose tissue.
Suzuki et al. [6] performed a meta-analysis of data from nine cohort and 22 case–control studies to examine the interrelationships between menopausal status, body weight and breast cancer ER and PR expression. In postmenopausal women, high body weight was associated with an 82 % increase in the risk for ER/PR-positive tumors; there was no relationship with ER-positive/PR-negative or ER/PR-negative tumors. In premenopausal women, there was a 20 % lower risk of developing ER/PR-positive tumors for those in the highest body weight category, a result that is consistent with the report by Cotterchio et al. [7] that in their case–control study of hormonal risk factors, obesity was associated with an increased risk of ER/PR-negative, and a reduced risk of ER/PR-positive breast cancer in premenopausal women. Also, Abrahamson et al. [27], in their study of obesity and survival in breast cancer, in which 82 % of the non-obese and 74 % of the obese were premenopausal, found that 62 % and 50 %, respectively, had ER-positive tumors (p = 0.005).
Triple-negative breast cancers are ER-, PR-, and HER-2-negative tumors of high histological grade, and highly metastatic (reviewed by [36]); they constitute approximately 10–15 % of breast cancers, with a higher prevalence in black women [37, 38]. There is a positive relationship between obesity and risk for triple-negative breast cancer which, in a meta-analysis based on 11 publications with a total of 24,479 breast cancer patients and 3,845 triple-negative tumors (15.7 %), was found by Pierobon & Frankenfeld [39] to be limited to premenopausal women. This relationship to premenopausal status was confirmed in a study of 1,884 patients from Turkey, 232 (12.3 %) of whom had triple-negative tumors [40].
Triple-negative breast cancer is also associated with social deprivation, although a complex interaction exists with obesity, race and ethnicity [41]. We performed a study of triple-negative breast cancer in white women in West Virginia, a state that has a 95 % White population, but ranks fourth in the country for the prevalence of obesity and sixth for the percent of the population that is below the poverty line [42]. Obesity was present in 49.6 % of those with triple-negative tumors but in only 35.8 % of those with other receptor combinations (p = 0.0098); the women with triple-negative tumors were also younger, with 44.5 % and 26.7 %, respectively being diagnosed at age <50 years (p = 0.0004).
Setiawan et al. [43] found that in their multiethnic study, 67 % of all postmenopausal patients, but 74 % of those who were obese, had breast cancers that were ER and PR-positive, and yet, despite these biomarkers of a good prognosis, the presence of obesity was associated with a poor clinical outcome. In explanation, it appears that ER positivity has only a weak, or no, modifying effect on an obesity-related poor prognosis [44, 45]. Enger et al. [44] observed a correlation between increasing body weight and breast cancer-specific mortality, and also an approximately 2-fold higher risk of dying from ER-negative compared with ER-positive cancer regardless of disease stage at the time of diagnosis. Niraula et al. [8] addressed the issue of hormone receptor status in their meta-analysis of the relationship between obesity and breast cancer prognosis; not only did they find that this was similar in premenopausal and postmenopausal patients, but there was no association with ER/PR status. These observations suggest the primacy of non-estrogen-related causes for the association of obesity in breast cancer with a poor clinical outcome. On the other hand, there is mounting evidence that obesity impairs the response to antiaromatase and tamoxifen therapy in ER-positive breast cancer. BMI significantly impacted the efficacy of anastrozole plus goserelin in premenopausal patients [46], and in postmenopausal women given tamoxifen plus aminoglutethimide [47].
Given that obesity is a risk factor for triple-negative breast cancer, it is counterintuitive that it has no effect on prognosis. However, a study of data from three adjuvant therapy trials by Sparano et al. [48] found that the adverse effect of obesity on disease-free and overall survivals applied specifically to patients with ER/PR-positive/HER-2-negative tumors and not to HER-2-overexpressing or triple-negative breast cancers. Dawood et al. [49] carried out a retrospective study of 2,311 patients with stage I to III triple-negative breast cancer, 35.7 % of whom were obese, and found no effect of BMI on distant disease-free survival, which they considered was due to the overwhelming adverse influence of the triple-negative phenotype, and a similar phenomenon may have been responsible in the case of HER-2-overexpressing tumors which are also associated with a poor clinical outcome.
Tumor Size, Proliferation Rates and Lymph Node Status
Obesity in newly diagnosed breast cancer patients is associated with pathological predictors of a poor prognosis, including large tumor size [48, 50–52], high histological grade [51, 52], and metastasis to the axillary lymph nodes [52, 53]. These relationships are present in both premenopausal and postmenopausal patients. Although the presence of more advanced disease could arise from delayed diagnosis in obese women, the evidence is more in favor of an association of obesity with aggressive, rapidly growing, tumors [54]. In addition to the high histologic grade, Daling et al. [51] found that breast cancers from obese women have a high S-phase fraction, mitotic cell count, and Ki-67 expression. Ki-67 is a nuclear proliferative biomarker that has been associated with a poor breast cancer prognosis [55], and was also shown to be overexpressed in the overweight and obese women with predominantly stage I breast cancers studied by Kamineni et al. [24] In a further analysis of their data, Daling et al. [51] showed that tumors with a ≥2.0 cm maximum diameter from women in the highest BMI quartile had higher levels of all three proliferative markers than tumors of the same size from women in the lowest BMI quartile, indicating that the large tumors in the overweight and obese women had been growing at a faster rate those from lean women.
Vascular Invasion and Distant Metastasis
The detection of lymphatic and vascular vessel invasion in primary breast cancers is strongly associated with tumor size, histologic grade and axillary lymph node involvement, but is also an independent predictor of local recurrence and impaired survival [56], and distant metastasis [57]. Badwe et al. [58] reported a positive association of vascular invasion with body size in postmenopausal breast cancer patients. In their study, vascular invasion was present in 36 % of cases; the median weight for those with vascular invasion was 69 kg and for those without it was 63 kg (p < 0.0001). The 5-year survival was significantly worse in the women with tumor vascular invasion (p < 0.0001). Several later studies demonstrated the increased prevalence of tumor vascular invasion in both postmenopausal and premenopausal obese breast cancers patients [59–61].
Obese breast cancer patients are more likely to have distant metastases. A prospective study in which the patients had been deliberately selected for early age at onset, so that 74 % were premenopausal, was reported from Australia by Loi et al. [62]. Obesity was present in 12 % of the 1,101 women, and 25 % were overweight. Distant metastases developed in 264 patients and this was related to obesity (HR 1.50; 95 % CI, 1.07-2.09; p = 0.02); although the obese women were more likely to have large primary tumors and axillary lymph node involvement at the time of diagnosis, obesity remained an independent predictor of distant recurrence after adjustment for these prognostic markers.
von Drygalski et al. [63] found that not only was obesity associated with more advanced disease at the time of the initial diagnosis, but that it had an adverse effect on survival once metastases were present, and was an independent predictor of time to metastasis and progression. In this important study, both progression-free (PFS) and overall survival (OS) were shortened by the presence of obesity: the median PFS for patients with a BMI ≤ 30 was 4.4 years and for a BMI >30 kg/m2 it was 2.5 years (p = 0.001); the corresponding median OS times were 7.1 and 3.2 years (p = 0.001). Once metastasis had been diagnosed, the median PFS for the obese and non-obese patients were 1.8 and 1.04 years, respectively and for OS they were 3.20 and 2.30 years (p < 0.02).
Obesity, Estrogens and Breast Cancer
Figure 1 summarizes the steps in the production of estrogens in adipose tissue and the regulation of their bioactivity. After the menopause, estrogens are produced almost exclusively in the stromal cells (preadipocytes) of the adipose tissue by the enzymatic aromatization of the C19 steroid androstenedione to form estrone; there is very little aromatase activity in the mature adipocytes [17]. Aromatase is the rate-limiting enzyme for estrogen synthesis, but adipose tissue also contains the 17β-hydroxysteroid dehydrogenase responsible for the conversion of estrone to the more biologically potent estradiol.

Synthesis of estrogens from the C19 steroid androstenedione in adipose tissue and regulation of estradiol bioavailability by sex hormone-binding globulin (SHBG). Androstenedione production by the adrenal glands is elevated in obesity, as is the activity of aromatase, an enzyme that is inducible by tumor necrosis factor-alpha (TNF-α) and prostaglandin E2 (PGE2)
Transcriptional regulation of the CYP19 gene that encodes for aromatase expression is tissue-specific due to several different promoters upstream that are activated by hormones, cytokines and PGE2. Zhao et al. [64] showed that TNF-α upregulates aromatase expression in adipose stromal cells by stimulating the binding of c-fos and c-jun transcription factors to an activating protein-1 (AP-1) binding site located upstream of promoter PI.4; PGE2 induces aromatase gene expression in the same cells via promoters PI.3/PII and a cAMP response element [65]. The recruitment of macrophages into adipose tissue provides a source of TNF-α and PGE2.
In breast cancer, aromatase expression in adipose stromal cells is elevated at locations adjacent to the tumor mass, an observation first made by O’Neill et al. [66] who found that activity of the enzyme was highest in adipose tissue from the cancer-containing quadrant of mastectomy specimens. Two possible explanations, not mutually exclusive, are that the breast cancer develops at a site, perhaps one of chronic low grade inflammation, that provides a local environment conducive to tumorigenesis (Fig. 2a), and that the cancer epithelial cells secrete paracrine factors that act on the stromal cells to induce aromatase expression (Fig. 2b). Bulun & Simpson [67] showed that not only was aromatase expression highest in the breast quadrant that contained the tumor, but that it also contained the highest proportion of adipose stromal cells (preadipocytes). This may be the result of the demonstrated inhibition of preadipocyte maturation by TNF-α [68] and/or dedifferentiation of adipocytes into preadipocytes [69–71], together with the reactivation of aromatase expression. There are two sources of this local TNF-α production: the tumor-associated macrophages (Fig. 2a) and the breast cancer cells themselves (Fig. 2b).

The local production of estrogens by preadipocytes (stromal cells) is modulated by changes in aromatase activity. As preadipocytes mature, enzyme expression is lost. a Tumor necrosis factor-alpha (TNF-α) and eicosanoids produced by infiltrating macrophages induce preadipocyte aromatase activity. Also, TNF-α acts on preadipocytes to arrest differentiation to mature adipocytes (left) and on adipocytes to stimulate their dedifferentiation back to preadiocytes (right). In both cases, the net effect is an increase in aromatase activity and estrogen production. b Similar stimulatory effects on aromatase activity may arise in a paracrine manner from TNF-α and prostaglandin E2 (PGE2) secreted by breast cancer cells
Postmenopausal plasma estrogen concentrations are elevated in obesity [72], but before the menopause they are unchanged in obese women because the dominant source is the ovaries. Obesity not only increases estradiol production in postmenopausal women, but also causes an elevation in its bioavailability due to a reduction in the synthesis of sex hormone-binding globulin (SHBG) by the liver. Under normal conditions approximately 30–40 % of the plasma estradiol is tightly bound to the SHBG and in consequence is rendered biologically inert. Most of the remainder is weakly bound to albumin, from which it is readily released, and another 1–2 % circulates as unbound ‘free’ estradiol; both of these fractions are available for biological activity.
The estrogens synthesized in the adipose tissues are generally accepted as having a causal role in breast cancer, and a meta-analysis of previously published studies by Key et al. [73] found that breast cancer risk in postmenopausal women increased with increasing concentrations of total plasma estrogens and the biologically available fraction (Fig. 1). Low plasma SHBG levels have also been associated with an increased breast cancer risk in postmenopausal women [73, 74]. Moreover, a positive association was found between postmenopausal breast cancer risk, increasing BMI, and high plasma estrogen and low SHBG concentrations [75].
Although these epidemiological observations are persuasive, they do not really distinguish between an endocrine mechanism, a consequence of a general production of estrogens in the elevated body fat mass, the commonly stated assumption, and paracrine interaction between the estrogen formed locally by aromatase activity in the breast adipose stromal cells and ER-positive target breast cancer cells (Fig. 2). In support of the latter view, Simpson & Davis [76] pointed out that tumor estrogen levels are considerably higher than those in plasma from postmenopausal women, and so any increase in the circulating concentrations may simply be a reflection of there being elevated local production in the breast. Also, in an aromatase transgenic mouse model, overexpression of the enzyme was able to sustain breast hyperplasia in the absence of circulating estrogen, an effect that was blocked by a pharmacological aromatase inhibitor [77].
Inflammation
Chronic inflammation is a prolonged condition in which tissue injury and attempts at repair coexist. In obese women a chronic low-grade inflammatory process occurs which is evidenced by an increase in the circulating levels of C-reactive protein (CRP), inflammatory cytokines such as TNF-α and interleukin (IL)-6, the chemokines IL-8 and monocyte chemoattractant protein-1 (MCP-1), and the mulifactorial and proinflammatory adipokine leptin [78]. With the exception of leptin, the principal source of these factors in obesity is the macrophages that have infiltrated the adipose tissue [79, 80].
Leptin is a product of the obese (ob) gene and is synthesized primarily in the preadipocytes and mature adipocytes; its production rate in the adipose tissue is directly proportional to the degree of adiposity and the plasma concentrations in healthy women are positively correlated with the BMI (reviewed by Vona-Davis & Rose [18]). Leptin has multiple functions, among which is its role in the modulation of innate and adaptive immune responses, with stimulation of monocyte proliferation and the production of TNF-α and other cytokines [81]. Like the proinflammatory cytokines with which it is associated, leptin promotes and maintains the low-grade inflammation associated with obesity.
In a review of the contributions of leptin to breast cancer risk and progression published in 2007, we concluded that the dominant mechanism by which leptin stimulates tumor cell growth, invasion and metastasis involves paracrine interaction with the cells of the surrounding adipose tissue; less clear was the role of obesity-related hyperleptinemia [18]. More recently, Ollberding et al. [82] reported the results of a nested case–control study which showed that although higher circulating levels of leptin were associated with increased postmenopausal breast cancer risk, the relationship was unaffected by adjustment for the BMI. Also, when Llanos et al. [83] compared the plasma and breast tissue leptin levels in women without a cancer history, they did find a positive correlation between the two, however the relationship was lost after adjustment for the BMI. One explanation for these inconsistent results is the possibility discussed earlier, that inflammation of the breast adipose tissue, rather than adiposity per se is the important etiologic factor.
Adiponectin is also an adipokine, but it has many functions that counter those of leptin: of particular significance to the present discussion, it is anti-inflammatory and inhibits TNF-α expression in macrophages and adipocytes [84]. The biological actions of adiponectin on breast cancer cells are in direct opposition to those of leptin, and hypoadiponectinemia, which occurs in obese women and chronic inflammatory states, has been associated with increased postmenopausal breast cancer risk and expression of an aggressive, metastatic, phenotype [18, 85]. Gross et al. [86] brought these features together in a case–control study that showed that high levels of plasma leptin and soluble TNF receptor 2, an inflammatory marker, but low levels of adiponectin, were associated with increased risk for postmenopausal breast cancer.
In obesity, the stroma vascular fraction of white adipose tissue is enriched in macrophages. In lean individuals, there are solitary “alternative” adipose tissue macrophages (M2) that are characterized by the expression of anti-inflammatory IL-10 and arginase to facilitate adipogenesis (Fig. 3). However, in obesity there are more “crown” aggregate macrophages (M1) found that preferentially express the surface marker integrin CD11c [87]. These pro-inflammatory “classical” macrophages are frequently associated with insulin resistance and obesity. Cancello et al. [88] found that after severe weight loss surgery, there were fewer crown-like structures within adipose tissue. The phenotypic shift from M1 to M2 could be viewed as an improvement in the inflammatory profile of the adipose tissue in human adipose tissue.

COX-2 and human macrophage differentiation in obesity and breast cancer. The anti-tumoral classically activated M1 macrophages are polarized by lipopolysaccharide (LPS), interferon gamma (IFN-γ) and tumor necrosis factor-alpha (TNF-α) to secrete proinflammatory cytokines and effector molecules. The pro-tumoral alternatively activated M2 macrophages become polarized by interleukin-10 (IL-10) and express anti-inflammatory molecules such as transforming growth factor-beta (TGF-β) and Arginase 1. In obesity, aggregate “crown” M1 macrophages are usually found in adipose tissue and associated with insulin resistance. In tumors, infiltrated macrophages are polarized by the M2 phenotype which promotes tumor growth. Cyclooxgenase-2 (COX-2) signaling during differentiation is required for M2 macrophage development; inhibiting COX-2 reduces their polarization and inhibits tumor promotion shifting monocyte differentiation toward M1 phenotype and systemic innate immunity
In cancer, the hormone-cytokine-eicosanoid interactions regulate both host cell and tumor cell response to that microenvironment. Tumor-associated macrophages (TAMs) predominately exhibit an M2 phenotype (Fig. 3). COX-2 is the key enzyme for differentiation of monocytes into alternatively activated M2 macrophages. Studies have shown that COX-2 inhibition alters the phenotype of tumor-associated macrophages from M2 to M1 in Apc min/+ mouse polyps [89]. This was recently confirmed by Na et al. [90] who showed that nonsteroidal anti-inflammatory drug therapy suppressed lung metastasis by reducing the M2 macrophage characteristics of TAMs in a murine model of breast cancer. Thus, it appears that blocking COX-2 may have clinical use in breast cancer as a therapy for induction of enhanced anti-tumor immunity in the prevention of breast cancer metastasis.
The inflammatory process may precede tumorigenesis, and this is the most likely situation in obesity-related breast cancer pathogenesis, but neoplastic transformation may also initiate the creation of a tumor-supporting inflammatory milieu [91]. It has been estimated that at least 20 % of all cancers originate in association with inflammation [92]. The prototypical proinflammatory cytokine is TNF-α, which is produced in the M1 or “classically activated” macrophages and is associated with adipocytic inflammation, angiogenesis in adipose tissue, and insulin resistance (reviewed by Shah et al. [93]); also, although originally described for its antitumor effects when present at high concentrations, it is now recognized as having multiple functions in promoting tumor development [91, 92].
A histological feature of adipose tissue inflammation is the aggregation of macrophages around individual adipocytes that consequently undergo necrosis and fuse to form a syncytium of lipid-containing giant multinucleated cells, referred to as “crown-like structures” [94]. Subbaramaiah et al. [9] found in obese mice that an increase in the number of crown-like structures in the stromal component of both the abdominal visceral fat and the mammary tissue was associated with an elevation in TNF-α and IL-6 production, induction of COX-2 and elevated PGE2 levels, and a corresponding increase in aromatase mRNA expression and enzyme activity. The macrophage-rich stromal-vascular fraction isolated from the mammary glands of obese mice secreted high levels of TNF-α, IL-1β, and PGE2 compared with the stromal-vascular fraction from lean mice, and by the use of blocking antibodies and a selective COX-2 inhibitor each fraction was shown to contribute to the induction of aromatase in preadipocytes. Moreover, treatment of macrophages with a saturated fatty acid stimulated production of the two cytokines and PGE2, and medium conditioned by the inflammatory cells induced preadipocyte aromatase expression; there were also elevations in cAMP and protein kinase A activities, known elements of the signaling pathway for PGE2 stimulation of CYP19 transcription. These products, including the estrogens synthesized by the increased aromatase activity, would then be available for paracrine stimulation of breast cancer cells (Fig. 4).

Obesity-related adipose tissue inflammation, infiltrating macrophages and stimulation of untransformed and/or transformed breast epithelial cells and vascular endothelial cells
The same research group also reported the association of crown-like structure accumulation and elevation of aromatase expression with higher BMI in non-cancerous breast adipose tissue from premenopausal and postmenopausal women with breast cancer [10]. The statistical correlation between the level of aromatase expression and the number of crown-like structures was stronger than that for the BMI, which the investigators pointed out is consistent with only a subset of postmenopausal obese women, those with breast adipose tissue inflammation and a high rate of local estrogen biosynthesis, being at increased breast cancer risk. This observation is consistent with the concept of “metabolically benign obesity”, which is characterized by an absence of insulin resistance [95], no increase in TNF-α or CRP expression [96], and a lack of crown-like structures [97]. There is an obvious need for epidemiological studies of obesity as a risk factor for breast cancer in which the focus is not on the BMI, which may not reflect the significant metabolic and cellular changes, but inflammation as indicated by crown-like structures, molecular markers of inflammation, and aromatase expression.
Cancer cells can lose their epithelial characteristics and assume a spindle-like or, fibroblastoid, mesenchymal morphology. This epithelial-mesenchymal transition (EMT) is marked by overexpression of the structural intermediate filament protein vimentin and downregulation of E-cadherin expression and results in increased migratory and invasive capacity [98] and a particularly aggressive phenotype in triple-negative breast cancer [99]. In addition to increasing extraglandular estrogen production by way of aromatase induction, obesity/inflammation-associated TNF-α may induce COX-2 and also has direct effects on breast cancer cells that contribute to EMT (Fig. 4), with elevated metalloproteinase expression, enhanced migration and invasive capacity [100–102], and metastasis [103]. Dunlap et al. [104] found that in a mouse model of diet-induced obesity promoted the development of mammary tumors with a mesenchymal phenotype, which exhibited upregulation of the EMT markers N-cadherin and Snail. Several studies have shown that adipocyte-conditioned medium and sera from obese rodents, which contain high concentrations of TNF-α and IL-6, can promote cancer cell EMT [105–107], and in untransformed MCF-10A breast epithelial cells, treatment with a combination of TNF-α and transforming growth factor-β induced a loss of E-cadherin and increased vimentin expression and enhanced migratory capacity [108].
Nuclear factor-κ light chain enhancer of activated B cells (NF-κB) is a transcription factor that regulates many of the genes implicated in tumor cell proliferation, migration and metastasis [109]. NF-κB also has a key role in chronic inflammation and activates the expression of genes encoding for inflammatory cytokines, COX-2, adhesion molecules and angiogenic factors. Most solid tumors and lymphoid malignancies possess activated NF-κB, which may be the result of mutational activation of upstream components of the signaling pathways or proinflammatory stimuli in the tumor microenvironment [110]. In co-culture experiments, macrophages were shown to increase the invasive capacity of breast cancer cells in a NF-κ-B and TNF-α-dependent manner [111].
Activation of NF-κB is regulated by interaction with “inhibitor of κB” (IκB) proteins; several such interactions have been described and involve different NF-κB and IκB proteins [112]. Proinflammatory molecules such as TNF-α activate NF-κB, so triggering series of events in the inflammatory process and mediating related stress responses [113, 114]. In the study of obesity and inflammation by Le et al. [97], the combined presence of crown-like structures and increased levels of TNF-α in adipose tissue was associated with upregulation of several genes of the NF-κB pathway.
In breast cancer, constitutively activated NF-κB was shown to be a downstream mediator of growth signaling in aggressively metastatic ER-negative, HER-2-positive [115, 116], and basal-like, triple-negative, breast cancer cells [117, 118]. In the studies by Dannenberg, Subbaramaiah and their colleagues discussed earlier [9, 10], the presence of crown-like structures in mammary adipose tissue was associated with activation of NF-κB that was involved in the upregulation of TNF-α, IL-1β and COX-2 in the macrophages, leading, in turn, to the induction of aromatase in stromal cells. Thus, obesity-related inflammation in breast cancer patients may also stimulate ER-positive breast cancer growth and progression by an NF-κB-related mechanism of increased local estrogen production.
Eicosanoids
The eicosanoids are autacoid mediators derived from a 20-carbon (eicosa-) omega-6 polyunsaturated fatty acid, arachidonic acid (5,8,11,14-eicosatetraenoic acid), and function on a local level as chemical transmitters for a variety of intracellular and intercellular signals. Arachidonic acid, produced in the liver from dietary linoleic acid, is stored as a constituent of the cell membrane phospholipids from which it is released under the influence of phospholipase A2. It provides the substrate for the two classes of enzymes with which we are concerned here: the PG synthases, usually referred to as COX, which produce the prostanoids (PGs, prostacyclin, and thromboxanes), and the lipoxygenases (LOXs), which catalyze the biosynthesis of the HETEs and LTs; the third class, comprising ω-hydroxylases and epoxygenases, form the cytochrome P450-derived eicosanoids (Fig. 5). There are two isoforms of COX: one, COX-1, is constitutively expressed by most tissues and is responsible for the generation of the PGs involved in physiological activities; the other, COX-2, is induced in a cell type-specific manner by various stimuli, including growth factors, and cytokines, notably TNF-α [119, 120].

The biosynthesis of cyclooxygenase and lipoxygenase products from arachidonic acid. Linoleic and arachidonic acid are both omega polyunsaturated fatty acids: linoleic acid is obtained entirely in the diet, whereas most arachidonic acid is formed in the liver by a series of desaturase and elongase reactions and stored in cell membrane phospholipid
The 5-, 12-, and 15-LOXs insert molecular oxygen into polyunsaturated fatty acid to form hydroperoxyeicosatetraenoic acids that are reduced to the hydroxyl analogues: 5-, 12-, and 15-HETE. The HETEs are involved in a wide range of biological activities, including the modulation of ion transport, various aspects of vascular, pulmonary and renal function, hormone secretion and the immune response.
Studies of eicosanoids in chronic inflammation have concentrated on the PGs, although more recently it has been recognized that the products of LOX-mediated pathways also play an important role [121, 122]. The adipose tissue-infiltrating macrophages express COX-2 [9, 11, 12] and LOXs [13, 123], and, in addition to PGE2, produce 5-, 12-, and 15-HETE and LT C4 [122].
Ristimaki et al. [124] examined the expression of COX-2 in 1,576 invasive breast cancers and found elevated levels of the enzyme protein in 37.4 %; these were associated with large tumor size and high proliferation rates, and with short distant disease-free survival. A similar study by Denkert et al. [125] showed a positive correlation between COX-2 expression and primary tumor size, axillary lymph node involvement, and poor histological differentiation; COX-2 overexpression was also related to reduced disease-free and overall survival. In a second publication by these investigators, data from eight different studies were reviewed; overexpression of COX-2 was present in 40 % of 2,392 tumors and was associated with pathological indicators of a poor prognosis and reduced survival [126]. There have been several reports that an inverse relationship exists between COX-2 and ER/PR expression in breast cancer tissues [124, 125, 127, 128], an observation that was not confirmed by Dhakal et al. [129], but is consistent with the association of COX-2-overexpressing and ER-negative tumors with biologically aggressive disease.
There are two proposed general mechanisms by which eicosanoids influence breast cancer growth, invasion and metastasis: one involves the PGE2-mediated induction of aromatase which has been discussed earlier in the review and applies only to ER-positive breast cancers; the other concerns the direct stimulatory effects of PGs, notably PGE2, and lipoxygenase products, HETEs and LTs, on growth and invasive/metastatic capacity, which is applicable to estrogen-independent, usually ER-negative, tumors which typically possess a more aggressive phenotype. However, the situation appears to be more complex: it has also been proposed that the local production of estrogens and proinflammatory cytokines which occurs in association with breast adipose tissue inflammation, with consequent activation of ER and NF-κB, promotes a more aggressive, but ER-positive, phenotype, and provides a mechanism by which breast cancers develop resistance to selective ER modulator and aromatase inhibitor therapy [130]. Moreover, Lykkesfeldt et al. [131] reported that although COX-2 expression may be associated with ER-positive breast cancer it does not predict responsiveness to endocrine therapy.
Although obesity-associated breast tissue inflammation may be accompanied by COX-2-related local estrogen production, as indicated by the work of Subbaramaiah et al. [12], and implied stimulation of tumorigenesis, there is also evidence from transfection experiments that COX-2 expression in breast cancer is associated with EMT, an accompanying loss of ER expression and emergence of the metastatic phenotype [132]. Also, arachidonic acid, the substrate for COX-2 activity, was shown to promote EMT of a human breast epithelial cell line [133]. This is clearly an important but complex area and merits further investigation.
There is substantial experimental evidence that LOX products are involved in breast cancer cell growth and expression of the metastatic phenotype. Early experiments with human ER-negative breast cancer cell lines and pharmacological inhibitors of eicosanoid synthesizing enzymes found that PGs did not stimulate proliferation in vitro [134, 135], but that 12-HETE was mitogenic and 12-LOX overexpression in the ER-positive MCF-7 breast cancer cell line produced an exaggerated growth response to linoleic acid [136]. MCF-7 cells transfected to overexpress 12-LOX also exhibited accelerated growth in athymic nude mice, with both increased proliferation and reduced apoptotic cell death, and high angiogenic activity [137]. More recently, Avis et al. [138] and Tong et al. [139] showed that 5-HETE and 12-HETE stimulate proliferation of several ER-positive and ER-negative breast cancer cell lines, and suppress apoptosis by upregulation of the anti-apoptotic gene Bcl-2 and blocking caspase-9 activation.
The level of 12-LOX mRNA expression has been found to be higher in breast cancer tissue than in uninvolved tissue from the same patient [128, 140, 141]. Mohammad et al. [128] reported that in addition to elevated COX-2 mRNA expression in 47 % of a small series of breast cancers, 63 % overexpressed 12-LOX; both were associated with higher tumor stage, but only COX-2 showed a negative relationship with ER expression and there was no correlation between COX-2 and 12-LOX.
The potential interactions between cytokines secreted by the inflammatory macrophages and breast cancer epithelial cells and eicosanoids in stimulating tumor growth and invasion are complex. Breast cancer cells possess varying levels of PG receptors, of which one, EP4, appears to have a key role in experimental metastasis [142], and Guo et al. [143] have identified a G protein-coupled transmembrane receptor for 12-HETE, and so the obesity-associated macrophages which infiltrate the adjacent adipose tissue may establish paracrine-autocrine loops involving macrophage cytokine-to-tumor cell COX/LOX and/or macrophage PGE2/HETE-to-tumor cell surface eicosanoid receptors.
Both COX-2 and lipoxygenase products also stimulate breast cancer cell invasion [144–147]. Serna-Marquez et al. [147] showed that linoleic acid stimulated breast cancer cell migration by a mechanism that involved the activation of focal adhesion kinase by a COX-2- and LOX-mediated process. In a series of experiments, Singh et al. [148] transfected MDA-MB-231 breast cancer cells so as to overexpress COX-2 and secrete high levels of PGE2; the observed increase in invasiveness was associated with elevated levels of urokinase-like plasminogen activator, a key proteolytic enzyme in the invasive process.
The catabolism of PGE2 in the body results in the production of a stable end-metabolite, PGE-M, which has been used an index of systemic PGE2 levels. Increased levels of urinary PGE-M occur in association with obesity, aging and lung metastases in patients with breast cancer [149]. As a biomarker of inflammation, PGE-M was positively associated with breast cancer risk in postmenopausal women who did not regularly use nonsteroidal anti-inflammatory drugs [150].
Angiogenesis and Obesity
Angiogenesis is a critical part of tumorigenesis and also facilitates spread to distant metastatic sites. A high level of neovascularization in primary breast cancer is predictive of a poor prognosis [151].
There is a complex relationship between adipose tissue and angiogenic activity, with the capillary bed undergoing expansion and contraction in response to fluctuations in adiposity; adipogenesis is regulated by new blood vessel formation, which itself is driven by preadipocyte- and macrophage-secreted angiogenic factors, and blood vessel density is governed by adipocyte number and size [152, 153]. In general, vascular endothelial growth factor (VEGF) is the dominant stimulating factor for angiogenesis, and the concentration in human plasma is positively correlated with the BMI [154], but there are several others, including leptin, platelet-derived growth factor (PDGF) and TNF-α [155], and some of the arachidonic acid-derived eicosanoids [156]. Experiments in vitro showed that leptin stimulates vascular endothelial cell growth, promotes degradation of the extracellular matrix so as to facilitate the establishment of a vascular network, and enhances capillary tube formation; in vivo, it was found to promote angiogenesis with a potency that is equal to that of VEGF.
Obesity-associated inflammation is considered to be a consequence of adipose tissue hypoxia and the associated infiltration by macrophages provides a source of TNF-α, PDGF, PGE2, and other angiogenic factors, including some VEGF, which, together VEGF and leptin produced in the adipocytes, stimulate neovascularization [157]. In inflammation-related cancer this local angiogenesis may contribute to the creation of a microenvironment that is favorable to tumor growth and metastasis [158].
Some COX-2 and LOX products are potent angiogenic factors. The proinflammatory eicosanoid PGE2 has been known to stimulate blood vessel formation for over 30 years; more recently, Zhang et al. [159] found that it exerts its effects through the EP4 receptor and promotes tube formation by microvascular endothelial cells, a function that in their experiments was mediated via protein kinase A. Celecoxib, a selective inhibitor of COX-2, suppressed the growth of a COX-2 overexpressing human colorectal cancer xenograft in athymic nude mice, together with lymph node metastasis, VEGF expression and tumor-related angiogenesis [160], and Prosperi et al. [161] showed that when MCF-7 breast cancer cells were transfected to overexpress COX-2 there was an increase in tumor cell proliferation, which was blocked by celecoxib, and invasive capacity; also, the cells expressed high levels of two VEGF splice variants.
The role of 12-HETE in tumor-related angiogenesis has been established in both cancers of the prostate [162, 163] and breast [137, 156]. Connolly & Rose [137] showed that in addition to accelerated growth, 12-LOX-overexpressing MCF-7 cell solid tumors in nude mice exhibit a high level of angiogenic activity. The expression of 5-LOX and 5-HETE and LTB4 production in a chemically-induced rat mammary carcinoma has also been related to a high level of angiogenesis and inflammation [164].
It is evident from this discussion that a complex network of adipocyte- and macrophage-associated angiogenic factors exists, which may undergo enhanced activity in obesity-related inflammation and promote breast cancer development and metastasis. Further studies are required to investigate the clinical significance of these observations, the specific contributions of the eicosanoids to the process, and the molecular mechanisms by which they impose their influence on angiogenesis.
Commentary
It needs to be stressed that the epidemiological and clinical-based contents of this review are derived almost exclusively from studies of white non-Hispanic women performed in North America and Europe. The distinction is important: the established association of obesity with an increased postmenopausal breast cancer risk which has been so clearly established in these studies may not hold for African-American or Hispanic women [165].
The recognition that adipose tissue inflammation is a major contributor to a wide range of metabolic diseases is having a considerable influence on the way we perceive the associations of obesity with several cancers, including carcinoma of the breast. In this context, the report by Hyatt et al. [166] that greater amounts of intra-abdominal adipose tissue were present in white compared with African-American women and associated with higher concentrations of circulating markers of inflammation, including TNF-α and IL-6, and that serum adiponectin levels were inversely related to these two cytokines in whites but not African-Americans, is of interest.
Adipose tissue inflammation occurs in association with type 2 diabetes and the metabolic syndrome, both of which have also been identified as risk factors for breast cancer; however, understanding the relationships is complicated by the concurrent role of obesity, which is itself causally associated with insulin resistance and a risk factor for both metabolic disorders. Colditz et al. [167] determined that in their cohort study of 113,861 women, 98 % of the cases of diabetes were attributable to obesity, and there is clearly considerable overlap in the biochemical features of obesity and type 2 diabetes that may be ascribed an etiologic role in breast cancer development; for example, in both conditions there is hyperinsulinemia, hyperleptinemia and hypoadiponectinemia. However, observational prospective studies on insulin serum levels and breast cancer risk have obtained mixed results. The association between serum insulin concentrations and breast cancer risk was not evident in a recent meta-analysis performed on prospective studies by Autier et al. [168]. The authors speculate that an increased risk found by some studies may have been due to inadequate control for BMI alone, as adiposity is the main cause of, rather than, a consequence of hyperinsulinemia.
Elevated plasma total and biologically available estrogen and reduced SHBG concentrations also occur in type 2 diabetes, but the problem is to distinguish the contribution of insulin from those of leptin and the inflammatory cytokines in producing these changes. Nevertheless, despite the problem of residual confounding after adjustment in the statistical analyses, the general view is that type 2 diabetes is associated with a modest increase in postmenopausal breast cancer risk and has an adverse effect on prognosis that are independent of any coexisting obesity. Insulin does stimulate the proliferation of ER-positive breast cancer cell lines and suppresses apoptosis, promotes tumor cell migration and invasion, and promotes angiogenesis (reviewed in Rose & Vona-Davis [169]).
Further studies are needed to address the relationship of breast adipose tissue inflammation to estrogen dependence in breast cancer. The demonstration by Subbaramaiah and his colleagues that obesity-associated inflammation is associated with the induction of breast stromal cell aromatase activity, a consequence of elevated COX-2 expression and PGE2 synthesis in proinflammatory macrophages, may have relevance only for postmenopausal ER-positive breast cancers and more specifically, albeit the majority, those that are truly estrogen dependent. It may be that inflammation is responsible for both an increased risk of ER-positive breast cancer and failure to respond to endocrine therapy, as suggested by Baumgarten & Frasor [130]. However, the situation is more complex than this: not only have a number of epidemiological studies shown an inverse relationship between ER and COX-2 expression, but at a molecular level, overexpression of COX-2 is a feature of epithelial-mesenchymal transition and the acquisition of a metastatic phenotype, including loss of ER expression. To complicate matters further, in spite of their aggressive biological behavior, triple-negative breast cancer tissues acquired at surgery have been reported to express only very low levels of both COX-2 and TNF-α.
The effects of short-term dietary energy restriction and/or exercise and weight reduction on intermediate end-points such as the plasma and adipose tissue concentrations of estrogens, inflammatory markers such as CRP, TNF-α and IL-6, and leptin have been reported, and putatively favorable reductions demonstrated. However, the translation of epidemiological and experimental studies of obesity, inflammation and breast cancer to preventive and therapeutic interventions will be much more difficult, although we do start with the experience gained from the dietary low-fat clinical trials. Indeed, in the WINS trial of a low-fat intervention in postmenopausal patients with stage I or II breast cancer, a reduction of dietary fat intake to 20 % of total calories was accompanied by a 6 lb weight loss which was associated with a 25 % decrease in the risk of recurrence; the Women’s Healthy Eating and Living Study, which produced no change in body weight showed no benefit of a low-fat diet (for reference see [170]).
In addition to interventions based on dietary and physical exercise, our review of the inflammation-eicosanoid connection in breast cancer suggests that for both prevention and treatment the efficacy of antiaromatase drugs might be enhanced by their combination with a COX inhibitor, or, perhaps even better, a combination of COX and LOX inhibition.
Abbreviations
- AP-1:
-
activating protein-1
- BMI:
-
body mass index
- COX-2:
-
cyclooxygenase-2
- CRP:
-
C-reactive protein
- EMT:
-
epithelial-mesenchymal transition
- ER:
-
estrogen receptor
- HETEs:
-
hydroxyeicosatetraenoic acids
- LTs:
-
leukotrienes
- LOX:
-
lipoxygenase
- MCP-1:
-
monocyte chemoattractant protein-1
- OS:
-
overall survival
- PDGF:
-
platelet-derived growth factor
- PFS:
-
progression-free survival
- PG:
-
prostaglandin
- PR:
-
progesterone receptor
- SHBG:
-
sex hormone-binding globulin
- TAMs:
-
tumor-associated macrophages
- TNF-α:
-
tumor necrosis factor-alpha
- VEGF:
-
vascular endothelial growth factor
- WHR:
-
waist-to-hip circumference
References
Harvie M, Hooper L, Howell AH. Central obesity and breast cancer risk: a systematic review. Obes Rev. 2003;4:157–73.
Stephenson GD, Rose DP. Breast cancer and obesity: an update. Nutr Cancer. 2003;45:1–16.
Rose DP, Vona-Davis L. Interaction between menopausal status and obesity in affecting breast cancer risk. Maturitas. 2010;66:33–8.
Maccio A, Madeddu C. Obesity, inflammation, and postmenopausal breast cancer: therapeutic implications. Sci World J. 2011;11:2020–36.
Amadou A, Ferrari P, Muwonge R, et al. Overweight, obesity and risk of premenopausal breast cancer according to ethnicity: a systematic review and dose-response meta-analysis. Obes Rev. 2013.
Suzuki R, Orsini N, Saji S, Key TJ, Wolk A. Body weight and incidence of breast cancer defined by estrogen and progesterone receptor status—a meta-analysis. Int J Cancer. 2009;124:698–712.
Cotterchio M, Kreiger N, Theis B, Sloan M, Bahl S. Hormonal factors and the risk of breast cancer according to estrogen- and progesterone-receptor subgroup. Cancer Epidemiol Biomarkers Prev. 2003;12:1053–60.
Niraula S, Ocana A, Ennis M, Goodwin PJ. Body size and breast cancer prognosis in relation to hormone receptor and menopausal status: a meta-analysis. Breast Cancer Res Treat. 2012;134:769–81.
Subbaramaiah K, Howe LR, Bhardwaj P, et al. Obesity is associated with inflammation and elevated aromatase expression in the mouse mammary gland. Cancer Prev Res (Phila). 2011;4:329–46.
Morris PG, Hudis CA, Giri D, et al. Inflammation and increased aromatase expression occur in the breast tissue of obese women with breast cancer. Cancer Prev Res (Phila). 2011;4:1021–9.
Hsieh PS, Jin JS, Chiang CF, Chan PC, Chen CH, Shih KC. COX-2-mediated inflammation in fat is crucial for obesity-linked insulin resistance and fatty liver. Obesity (Silver Spring). 2009;17:1150–7.
Subbaramaiah K, Morris PG, Zhou XK, et al. Increased levels of COX-2 and prostaglandin E2 contribute to elevated aromatase expression in inflamed breast tissue of obese women. Cancer Discov. 2012;2:356–65.
Chakrabarti SK, Wen Y, Dobrian AD, et al. Evidence for activation of inflammatory lipoxygenase pathways in visceral adipose tissue of obese Zucker rats. Am J Physiol Endocrinol Metab. 2011;300:E175–87.
Flegal KM, Carroll MD, Kit BK, Ogden CL. Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999–2010. JAMA. 2012;307:491–7.
Rodriguez A, Catalan V, Gomez-Ambrosi J, Fruhbeck G. Visceral and subcutaneous adiposity: are both potential therapeutic targets for tackling the metabolic syndrome? Curr Pharm Des. 2007;13:2169–75.
Alvehus M, Buren J, Sjostrom M, Goedecke J, Olsson T. The human visceral fat depot has a unique inflammatory profile. Obesity (Silver Spring). 2010;18:879–83.
Bulun SE, Chen D, Moy I, Brooks DC, Zhao H. Aromatase, breast cancer and obesity: a complex interaction. Trends Endocrinol Metab. 2012;23:83–9.
Vona-Davis L, Rose DP. Adipokines as endocrine, paracrine, and autocrine factors in breast cancer risk and progression. Endocr Relat Cancer. 2007;14:189–206.
Friedenreich CM, Courneya KS, Bryant HE. Case-control study of anthropometric measures and breast cancer risk. Int J Cancer. 2002;99:445–52.
Connolly BS, Barnett C, Vogt KN, Li T, Stone J, Boyd NF. A meta-analysis of published literature on waist-to-hip ratio and risk of breast cancer. Nutr Cancer. 2002;44:127–38.
Huang Z, Willett WC, Colditz GA, et al. Waist circumference, waist:hip ratio, and risk of breast cancer in the Nurses’ Health Study. Am J Epidemiol. 1999;150:1316–24.
Carmichael AR. Obesity and prognosis of breast cancer. Obes Rev. 2006;7:333–40.
Protani M, Coory M, Martin JH. Effect of obesity on survival of women with breast cancer: systematic review and meta-analysis. Breast Cancer Res Treat. 2010;123:627–35.
Kamineni A, Anderson ML, White E, et al. Body mass index, tumor characteristics, and prognosis following diagnosis of early-stage breast cancer in a mammographically screened population. Cancer Causes Control. 2013;24:305–12.
Dignam JJ, Wieand K, Johnson KA, et al. Effects of obesity and race on prognosis in lymph node-negative, estrogen receptor-negative breast cancer. Breast Cancer Res Treat. 2006;97:245–54.
Borugian MJ, Sheps SB, Kim-Sing C, et al. Waist-to-hip ratio and breast cancer mortality. Am J Epidemiol. 2003;158:963–8.
Abrahamson PE, Gammon MD, Lund MJ, et al. General and abdominal obesity and survival among young women with breast cancer. Cancer Epidemiol Biomarkers Prev. 2006;15:1871–7.
Esteva FJ, Hortobagyi GN. Prognostic molecular markers in early breast cancer. Breast Cancer Res. 2004;6:109–18.
Cui X, Zhang P, Deng W, et al. Insulin-like growth factor-I inhibits progesterone receptor expression in breast cancer cells via the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin pathway: progesterone receptor as a potential indicator of growth factor activity in breast cancer. Mol Endocrinol. 2003;17:575–88.
Cui X, Schiff R, Arpino G, Osborne CK, Lee AV. Biology of progesterone receptor loss in breast cancer and its implications for endocrine therapy. J Clin Oncol. 2005;23:7721–35.
Banegas MP, Li CI. Breast cancer characteristics and outcomes among Hispanic Black and Hispanic White women. Breast Cancer Res Treat. 2012;134:1297–304.
Colditz GA, Rosner BA, Chen WY, Holmes MD, Hankinson SE. Risk factors for breast cancer according to estrogen and progesterone receptor status. J Natl Cancer Inst. 2004;96:218–28.
Huang WY, Newman B, Millikan RC, Schell MJ, Hulka BS, Moorman PG. Hormone-related factors and risk of breast cancer in relation to estrogen receptor and progesterone receptor status. Am J Epidemiol. 2000;151:703–14.
Enger SM, Ross RK, Paganini-Hill A, Carpenter CL, Bernstein L. Body size, physical activity, and breast cancer hormone receptor status: results from two case-control studies. Cancer Epidemiol Biomarkers Prev. 2000;9:681–7.
Rosenberg LU, Einarsdottir K, Friman EI, et al. Risk factors for hormone receptor-defined breast cancer in postmenopausal women. Cancer Epidemiol Biomarkers Prev. 2006;15:2482–8.
Reis-Filho JS, Tutt AN. Triple negative tumours: a critical review. Histopathology. 2008;52:108–18.
Morris GJ, Naidu S, Topham AK, et al. Differences in breast carcinoma characteristics in newly diagnosed African-American and Caucasian patients: a single-institution compilation compared with the National Cancer Institute’s Surveillance, Epidemiology, and End Results database. Cancer. 2007;110:876–84.
Jack RH, Davies EA, Renshaw C, et al. Differences in breast cancer hormone receptor status in ethnic groups: a London population. Eur J Cancer. 2013;49:696–702.
Pierobon M, Frankenfeld CL. Obesity as a risk factor for triple-negative breast cancers: a systematic review and meta-analysis. Breast Cancer Res Treat. 2013;137:307–14.
Turkoz FP, Solak M, Petekkaya I, et al. Association between common risk factors and molecular subtypes in breast cancer patients. Breast. 2013;22:344–50.
Vona-Davis L, Rose DP. The influence of socioeconomic disparities on breast cancer tumor biology and prognosis: a review. J Womens Health (Larchmt). 2009;18:883–93.
Vona-Davis L, Rose DP, Hazard H, et al. Triple-negative breast cancer and obesity in a rural Appalachian population. Cancer Epidemiol Biomarkers Prev. 2008;17:3319–24.
Setiawan VW, Monroe KR, Wilkens LR, Kolonel LN, Pike MC, Henderson BE. Breast cancer risk factors defined by estrogen and progesterone receptor status: the multiethnic cohort study. Am J Epidemiol. 2009;169:1251–9.
Enger SM, Greif JM, Polikoff J, Press M. Body weight correlates with mortality in early-stage breast cancer. Arch Surg. 2004;139:954–8.
Cleveland RJ, Eng SM, Abrahamson PE, et al. Weight gain prior to diagnosis and survival from breast cancer. Cancer Epidemiol Biomarkers Prev. 2007;16:1803–11.
Pfeiler G, Konigsberg R, Fesl C, et al. Impact of body mass index on the efficacy of endocrine therapy in premenopausal patients with breast cancer: an analysis of the prospective ABCSG-12 trial. J Clin Oncol. 2011;29:2653–9.
Pfeiler G, Stoger H, Dubsky P, et al. Efficacy of tamoxifen +/− aminoglutethimide in normal weight and overweight postmenopausal patients with hormone receptor-positive breast cancer: an analysis of 1509 patients of the ABCSG-06 trial. Br J Cancer. 2013;108:1408–14.
Sparano JA, Wang M, Zhao F, et al. Obesity at diagnosis is associated with inferior outcomes in hormone receptor-positive operable breast cancer. Cancer. 2012;118:5937–46.
Dawood S, Lei X, Litton JK, Buchholz TA, Hortobagyi GN, Gonzalez-Angulo AM. Impact of body mass index on survival outcome among women with early stage triple-negative breast cancer. Clin Breast Cancer. 2012;12:364–72.
Maehle BO, Tretli S, Skjaerven R, Thorsen T. Premorbid body weight and its relations to primary tumour diameter in breast cancer patients; its dependence on estrogen and progesteron receptor status. Breast Cancer Res Treat. 2001;68:159–69.
Daling JR, Malone KE, Doody DR, Johnson LG, Gralow JR, Porter PL. Relation of body mass index to tumor markers and survival among young women with invasive ductal breast carcinoma. Cancer. 2001;92:720–9.
Markkula A, Bromee A, Henningson M, et al. Given breast cancer, does breast size matter? Data from a prospective breast cancer cohort. Cancer Causes Control. 2012;23:1307–16.
Maehle BO, Tretli S, Thorsen T. The associations of obesity, lymph node status and prognosis in breast cancer patients: dependence on estrogen and progesterone receptor status. APMIS. 2004;112:349–57.
Rose DP, Vona-Davis L. Influence of obesity on breast cancer receptor status and prognosis. Expert Rev Anticancer Ther. 2009;9:1091–101.
de Azambuja E, Cardoso F, De Castro Jr G, et al. Ki-67 as prognostic marker in early breast cancer: a meta-analysis of published studies involving 12,155 patients. Br J Cancer. 2007;96:1504–13.
Pinder SE, Ellis IO, Galea M, O’Rouke S, Blamey RW, Elston CW. Pathological prognostic factors in breast cancer. III. Vascular invasion: relationship with recurrence and survival in a large study with long-term follow-up. Histopathology. 1994;24:41–7.
Westenend PJ, Meurs CJ, Damhuis RA. Tumour size and vascular invasion predict distant metastasis in stage I breast cancer. Grade distinguishes early and late metastasis. J Clin Pathol. 2005;58:196–201.
Badwe RA, Fentiman IS, Millis RR, Gregory WM. Body weight and vascular invasion in post-menopausal women with breast cancer. Br J Cancer. 1997;75:910–3.
Demirkan B, Alacacioglu A, Yilmaz U. Relation of body mass index (BMI) to disease free (DFS) and distant disease free survivals (DDFS) among Turkish women with operable breast carcinoma. Jpn J Clin Oncol. 2007;37:256–65.
Gillespie EF, Sorbero ME, Hanauer DA, et al. Obesity and angiolymphatic invasion in primary breast cancer. Ann Surg Oncol. 2010;17:752–9.
Biglia N, Peano E, Sgandurra P, et al. Body mass index (BMI) and breast cancer: impact on tumor histopatologic features, cancer subtypes and recurrence rate in pre and postmenopausal women. Gynecol Endocrinol. 2013;29:263–7.
Loi S, Milne RL, Friedlander ML, et al. Obesity and outcomes in premenopausal and postmenopausal breast cancer. Cancer Epidemiol Biomarkers Prev. 2005;14:1686–91.
von Drygalski A, Tran TB, Messer K, et al. Obesity is an independent predictor of poor survival in metastatic breast cancer: retrospective analysis of a patient cohort whose treatment included high-dose chemotherapy and autologous stem cell support. Int J Breast Cancer. 2011;2011:523276.
Zhao Y, Nichols JE, Valdez R, Mendelson CR, Simpson ER. Tumor necrosis factor-alpha stimulates aromatase gene expression in human adipose stromal cells through use of an activating protein-1 binding site upstream of promoter 1.4. Mol Endocrinol. 1996;10:1350–7.
Chen D, Reierstad S, Lin Z, et al. Prostaglandin E(2) induces breast cancer related aromatase promoters via activation of p38 and c-Jun NH(2)-terminal kinase in adipose fibroblasts. Cancer Res. 2007;67:8914–22.
O’Neill JS, Elton RA, Miller WR. Aromatase activity in adipose tissue from breast quadrants: a link with tumour site. Br Med J (Clin Res Ed). 1988;296:741–3.
Bulun SE, Simpson ER. Regulation of aromatase expression in human tissues. Breast Cancer Res Treat. 1994;30:19–29.
Sethi JK, Hotamisligil GS. The role of TNF alpha in adipocyte metabolism. Semin Cell Dev Biol. 1999;10:19–29.
Prins JB, Niesler CU, Winterford CM, et al. Tumor necrosis factor-alpha induces apoptosis of human adipose cells. Diabetes. 1997;46:1939–44.
Engelman JA, Berg AH, Lewis RY, Lisanti MP, Scherer PE. Tumor necrosis factor alpha-mediated insulin resistance, but not dedifferentiation, is abrogated by MEK1/2 inhibitors in 3T3-L1 adipocytes. Mol Endocrinol. 2000;14:1557–69.
Nieman KM, Romero IL, Van Houten B, Lengyel E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim.Biophys Acta. 2013.
Baglietto L, English DR, Hopper JL, et al. Circulating steroid hormone concentrations in postmenopausal women in relation to body size and composition. Breast Cancer Res Treat. 2009;115:171–9.
Key T, Appleby P, Barnes I, Reeves G, Endogenous Hormones and Breast Cancer Collaborative Group. Endogenous sex hormones and breast cancer in postmenopausal women: reanalysis of nine prospective studies. J Natl Cancer Inst. 2002;94:606–16.
Zeleniuch-Jacquotte A, Shore RE, Koenig KL, et al. Postmenopausal levels of oestrogen, androgen, and SHBG and breast cancer: long-term results of a prospective study. Br J Cancer. 2004;90:153–9.
Rinaldi S, Key TJ, Peeters PH, et al. Anthropometric measures, endogenous sex steroids and breast cancer risk in postmenopausal women: a study within the EPIC cohort. Int J Cancer. 2006;118:2832–9.
Simpson ER, Davis SR. Minireview: aromatase and the regulation of estrogen biosynthesis–some new perspectives. Endocrinology. 2001;142:4589–94.
Tekmal RR, Kirma N, Gill K, Fowler K. Aromatase overexpression and breast hyperplasia, an in vivo model–continued overexpression of aromatase is sufficient to maintain hyperplasia without circulating estrogens, and aromatase inhibitors abrogate these preneoplastic changes in mammary glands. Endocr Relat Cancer. 1999;6:307–14.
Zeyda M, Stulnig TM. Obesity, inflammation, and insulin resistance—a mini-review. Gerontology. 2009;55:379–86.
Fain JN, Madan AK, Hiler ML, Cheema P, Bahouth SW. Comparison of the release of adipokines by adipose tissue, adipose tissue matrix, and adipocytes from visceral and subcutaneous abdominal adipose tissues of obese humans. Endocrinology. 2004;145:2273–82.
Fain JN. Release of inflammatory mediators by human adipose tissue is enhanced in obesity and primarily by the nonfat cells: a review. Mediat Inflamm. 2010;2010:513948.
Iikuni N, Lam QL, Lu L, Matarese G, La Cava A. Leptin and Inflammation. Curr Immunol Rev. 2008;4:70–9.
Ollberding NJ, Kim Y, Shvetsov YB, et al. Prediagnostic leptin, adiponectin, C-reactive protein, and the risk of postmenopausal breast cancer. Cancer Prev Res (Phila). 2013;6:188–95.
Llanos AA, Dumitrescu RG, Marian C, et al. Adipokines in plasma and breast tissues: associations with breast cancer risk factors. Cancer Epidemiol Biomarkers Prev. 2012;21:1745–55.
Villarreal-Molina MT, Antuna-Puente B. Adiponectin: anti-inflammatory and cardioprotective effects. Biochimie. 2012;94:2143–9.
Grossmann ME, Ray A, Nkhata KJ, et al. Obesity and breast cancer: status of leptin and adiponectin in pathological processes. Cancer Metastasis Rev. 2010;29:641–53.
Gross A, Newschaffer CJ, Hoffman Bolton JA, Rifai N, Visvanathan K. Adipocytokines, inflammation, and breast cancer risk in postmenopausal women: A prospective study. Cancer Epidemiol Biomarkers Prev. 2013.
Wentworth JM, Naselli G, Brown WA, et al. Pro-inflammatory CD11c + CD206+ adipose tissue macrophages are associated with insulin resistance in human obesity. Diabetes. 2010;59:1648–56.
Cancello R, Henegar C, Viguerie N, et al. Reduction of macrophage infiltration and chemoattractant gene expression changes in white adipose tissue of morbidly obese subjects after surgery-induced weight loss. Diabetes. 2005;54:2277–86.
Nakanishi Y, Nakatsuji M, Seno H, et al. COX-2 inhibition alters the phenotype of tumor-associated macrophages from M2 to M1 in ApcMin/+ mouse polyps. Carcinogenesis. 2011;32:1333–9.
Na YR, Yoon YN, Son DI, Seok SH. Cyclooxygenase-2 inhibition blocks M2 macrophage differentiation and suppresses metastasis in murine breast cancer model. PLoS One. 2013;8:e63451.
Candido J, Hagemann T. Cancer-related inflammation. J Clin Immunol. 2013;33 Suppl 1:S79–84.
Grivennikov SI, Karin M. Inflammatory cytokines in cancer: tumour necrosis factor and interleukin 6 take the stage. Ann Rheum Dis. 2011;70 Suppl 1:i104–8.
Shah A, Mehta N, Reilly MP. Adipose inflammation, insulin resistance, and cardiovascular disease. JPEN J Parenter Enteral Nutr. 2008;32:638–44.
Cinti S, Mitchell G, Barbatelli G, et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res. 2005;46:2347–55.
Stefan N, Kantartzis K, Machann J, et al. Identification and characterization of metabolically benign obesity in humans. Arch Intern Med. 2008;168:1609–16.
Karelis AD, Faraj M, Bastard JP, et al. The metabolically healthy but obese individual presents a favorable inflammation profile. J Clin Endocrinol Metab. 2005;90:4145–50.
Le KA, Mahurkar S, Alderete TL, et al. Subcutaneous adipose tissue macrophage infiltration is associated with hepatic and visceral fat deposition, hyperinsulinemia, and stimulation of NF-kappaB stress pathway. Diabetes. 2011;60:2802–9.
Foroni C, Broggini M, Generali D, Damia G. Epithelial-mesenchymal transition and breast cancer: role, molecular mechanisms and clinical impact. Cancer Treat Rev. 2012;38:689–97.
Karihtala P, Auvinen P, Kauppila S, Haapasaari KM, Jukkola-Vuorinen A, Soini Y. Vimentin, zeb1 and Sip1 are up-regulated in triple-negative and basal-like breast cancers: association with an aggressive tumour phenotype. Breast Cancer Res Treat. 2013;138:81–90.
Kim S, Choi JH, Kim JB, et al. Berberine suppresses TNF-alpha-induced MMP-9 and cell invasion through inhibition of AP-1 activity in MDA-MB-231 human breast cancer cells. Molecules. 2008;13:2975–85.
Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM, Zhou BP. Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell. 2009;15:416–28.
Cho SG, Li D, Stafford LJ, et al. KiSS1 suppresses TNFalpha-induced breast cancer cell invasion via an inhibition of RhoA-mediated NF-kappaB activation. J Cell Biochem. 2009;107:1139–49.
Hamaguchi T, Wakabayashi H, Matsumine A, Sudo A, Uchida A. TNF inhibitor suppresses bone metastasis in a breast cancer cell line. Biochem Biophys Res Commun. 2011;407:525–30.
Dunlap SM, Chiao LJ, Nogueira L, et al. Dietary energy balance modulates epithelial-to-mesenchymal transition and tumor progression in murine claudin-low and basal-like mammary tumor models. Cancer Prev Res (Phila). 2012;5:930–42.
Kushiro K, Chu RA, Verma A, Nunez NP. Adipocytes promote B16BL6 melanoma cell invasion and the epithelial-to-Mesenchymal Transition. Cancer Microenviron. 2012;5:73–82.
Kushiro K, Nunez NP. Ob/ob serum promotes a mesenchymal cell phenotype in B16BL6 melanoma cells. Clin Exp Metastasis. 2011;28:877–86.
Price RS, Cavazos DA, De Angel RE, Hursting SD, deGraffenried LA. Obesity-related systemic factors promote an invasive phenotype in prostate cancer cells. Prostate Cancer Prostatic Dis. 2012;15:135–43.
Sehrawat A, Singh SV. Benzyl isothiocyanate inhibits epithelial-mesenchymal transition in cultured and xenografted human breast cancer cells. Cancer Prev Res (Phila). 2011;4:1107–17.
Van Waes C. Nuclear factor-kappaB in development, prevention, and therapy of cancer. Clin Cancer Res. 2007;13:1076–82.
Karin M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol. 2009;1:a000141.
Hagemann T, Wilson J, Kulbe H, et al. Macrophages induce invasiveness of epithelial cancer cells via NF-kappa B and JNK. J Immunol. 2005;175:1197–205.
Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–62.
Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–8.
Ahn KS, Aggarwal BB. Transcription factor NF-kappaB: a sensor for smoke and stress signals. Ann N Y Acad Sci. 2005;1056:218–33.
Singh S, Shi Q, Bailey ST, et al. Nuclear factor-kappaB activation: a molecular therapeutic target for estrogen receptor-negative and epidermal growth factor receptor family receptor-positive human breast cancer. Mol Cancer Ther. 2007;6:1973–82.
Ahmed A. Prognostic and therapeutic role of nuclear factor-kappa B (NF-kappaB) in breast cancer. J Ayub Med Coll Abbottabad. 2010;22:218–21.
Yamaguchi N, Ito T, Azuma S, et al. Constitutive activation of nuclear factor-kappaB is preferentially involved in the proliferation of basal-like subtype breast cancer cell lines. Cancer Sci. 2009;100:1668–74.
Sethi S, Sarkar FH, Ahmed Q, et al. Molecular markers of epithelial-to-mesenchymal transition are associated with tumor aggressiveness in breast carcinoma. Transl Oncol. 2011;4:222–6.
Newton R, Kuitert LM, Slater DM, Adcock IM, Barnes PJ. Cytokine induction of cytosolic phospholipase A2 and cyclooxygenase-2 mRNA is suppressed by glucocorticoids in human epithelial cells. Life Sci. 1997;60:67–78.
Hobbs SS, Goettel JA, Liang D, et al. TNF transactivation of EGFR stimulates cytoprotective COX-2 expression in gastrointestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2011;301:G220–9.
Gonzalez-Periz A, Claria J. Resolution of adipose tissue inflammation. Sci World J. 2010;10:832–56.
Long EK, Hellberg K, Foncea R, Hertzel AV, Suttles J, Bernlohr DA. Fatty acids induce leukotriene C4 synthesis in macrophages in a fatty acid binding protein-dependent manner. Biochim Biophys Acta. 1831;2013:1199–207.
Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294:1871–5.
Ristimaki A, Sivula A, Lundin J, et al. Prognostic significance of elevated cyclooxygenase-2 expression in breast cancer. Cancer Res. 2002;62:632–5.
Denkert C, Winzer KJ, Muller BM, et al. Elevated expression of cyclooxygenase-2 is a negative prognostic factor for disease free survival and overall survival in patients with breast carcinoma. Cancer. 2003;97:2978–87.
Denkert C, Winzer KJ, Hauptmann S. Prognostic impact of cyclooxygenase-2 in breast cancer. Clin Breast Cancer. 2004;4:428–33.
Kim HS, Moon HG, Han W, et al. COX2 overexpression is a prognostic marker for Stage III breast cancer. Breast Cancer Res Treat. 2012;132:51–9.
Mohammad AM, Abdel HA, Abdel W, Ahmed AM, Wael T, Eiman G. Expression of cyclooxygenase-2 and 12-lipoxygenase in human breast cancer and their relationship with HER-2/neu and hormonal receptors: impact on prognosis and therapy. Indian J Cancer. 2006;43:163–8.
Dhakal HP, Naume B, Synnestvedt M, et al. Expression of cyclooxygenase-2 in invasive breast carcinomas and its prognostic impact. Histol Histopathol. 2012;27:1315–25.
Baumgarten SC, Frasor J. Minireview: Inflammation: an instigator of more aggressive estrogen receptor (ER) positive breast cancers. Mol Endocrinol. 2012;26:360–71.
Lykkesfeldt AE, Henriksen KL, Rasmussen BB, et al. In situ aromatase expression in primary tumor is associated with estrogen receptor expression but is not predictive of response to endocrine therapy in advanced breast cancer. BMC Cancer. 2009;9:185.
Kang JH, Song KH, Jeong KC, et al. Involvement of Cox-2 in the metastatic potential of chemotherapy-resistant breast cancer cells. BMC Cancer. 2011;11:334.
Martinez-Orozco R, Navarro-Tito N, Soto-Guzman A, Castro-Sanchez L, Perez SE. Arachidonic acid promotes epithelial-to-mesenchymal-like transition in mammary epithelial cells MCF10A. Eur J Cell Biol. 2010;89:476–88.
Hughes R, Timmermans P, Schrey MP. Regulation of arachidonic acid metabolism, aromatase activity and growth in human breast cancer cells by interleukin-1beta and phorbol ester: dissociation of a mediatory role for prostaglandin E2 in the autocrine control of cell function. Int J Cancer. 1996;67:684–9.
Rose DP, Connolly JM. Effects of fatty acids and inhibitors of eicosanoid synthesis on the growth of a human breast cancer cell line in culture. Cancer Res. 1990;50:7139–44.
Liu XH, Connolly JM, Rose DP. The 12-lipoxygenase gene-transfected MCF-7 human breast cancer cell line exhibits estrogen-independent, but estrogen and omega-6 fatty acid-stimulated proliferation in vitro, and enhanced growth in athymic nude mice. Cancer Lett. 1996;109:223–30.
Connolly JM, Rose DP. Enhanced angiogenesis and growth of 12-lipoxygenase gene-transfected MCF-7 human breast cancer cells in athymic nude mice. Cancer Lett. 1998;132:107–12.
Avis I, Hong SH, Martinez A, et al. Five-lipoxygenase inhibitors can mediate apoptosis in human breast cancer cell lines through complex eicosanoid interactions. FASEB J. 2001;15:2007–9.
Tong WG, Ding XZ, Adrian TE. The mechanisms of lipoxygenase inhibitor-induced apoptosis in human breast cancer cells. Biochem Biophys Res Commun. 2002;296:942–8.
Natarajan R, Esworthy R, Bai W, Gu JL, Wilczynski S, Nadler J. Increased 12-lipoxygenase expression in breast cancer tissues and cells. Regulation by epidermal growth factor. J Clin Endocrinol Metab. 1997;82:1790–8.
Jiang WG, Douglas-Jones A, Mansel RE. Levels of expression of lipoxygenases and cyclooxygenase-2 in human breast cancer. Prostaglandins Leukot Essent Fat Acids. 2003;69:275–81.
Fulton AM, Ma X, Kundu N. Targeting prostaglandin E EP receptors to inhibit metastasis. Cancer Res. 2006;66:9794–7.
Guo Y, Zhang W, Giroux C, et al. Identification of the orphan G protein-coupled receptor GPR31 as a receptor for 12-(S)-hydroxyeicosatetraenoic acid. J Biol Chem. 2011;286:33832–40.
Liu XH, Connolly JM, Rose DP. Eicosanoids as mediators of linoleic acid-stimulated invasion and type IV collagenase production by a metastatic human breast cancer cell line. Clin Exp Metastasis. 1996;14:145–52.
Timoshenko AV, Xu G, Chakrabarti S, Lala PK, Chakraborty C. Role of prostaglandin E2 receptors in migration of murine and human breast cancer cells. Exp Cell Res. 2003;289:265–74.
Larkins TL, Nowell M, Singh S, Sanford GL. Inhibition of cyclooxygenase-2 decreases breast cancer cell motility, invasion and matrix metalloproteinase expression. BMC Cancer. 2006;6:181.
Serna-Marquez N, Villegas-Comonfort S, Galindo-Hernandez O, Navarro-Tito N, Millan A, Salazar EP. Role of LOXs and COX-2 on FAK activation and cell migration induced by linoleic acid in MDA-MB-231 breast cancer cells. Cell Oncol (Dordr). 2013;36:65–77.
Singh B, Berry JA, Shoher A, Ramakrishnan V, Lucci A. COX-2 overexpression increases motility and invasion of breast cancer cells. Int J Oncol. 2005;26:1393–9.
Morris PG, Zhou XK, Milne GL, et al. Increased levels of urinary PGE-M, a biomarker of inflammation, occur in association with obesity, aging, and lung metastases in patients with breast cancer. Cancer Prev Res (Phila). 2013;6:428–36.
Kim S, Taylor JA, Milne GL, Sandler DP. Association between urinary prostaglandin E2 metabolite and breast cancer risk: a prospective, Case-Cohort Study of Postmenopausal Women. Cancer Prev Res (Phila). 2013;6:511–8.
Uzzan B, Nicolas P, Cucherat M, Perret GY. Microvessel density as a prognostic factor in women with breast cancer: a systematic review of the literature and meta-analysis. Cancer Res. 2004;64:2941–55.
Pang C, Gao Z, Yin J, Zhang J, Jia W, Ye J. Macrophage infiltration into adipose tissue may promote angiogenesis for adipose tissue remodeling in obesity. Am J Physiol Endocrinol Metab. 2008;295:E313–22.
Christiaens V, Lijnen HR. Angiogenesis and development of adipose tissue. Mol Cell Endocrinol. 2010;318:2–9.
Wada H, Ura S, Kitaoka S, et al. Distinct characteristics of circulating vascular endothelial growth factor-a and C levels in human subjects. PLoS One. 2011;6:e29351.
Vona-Davis L, Rose DP. Angiogenesis, adipokines and breast cancer. Cytokine Growth Factor Rev. 2009;20:193–201.
Rose DP, Connolly JM. Regulation of tumor angiogenesis by dietary fatty acids and eicosanoids. Nutr Cancer. 2000;37:119–27.
Ye J. Adipose tissue vascularization: its role in chronic inflammation. Curr Diab Rep. 2011;11:203–10.
Ono M. Molecular links between tumor angiogenesis and inflammation: inflammatory stimuli of macrophages and cancer cells as targets for therapeutic strategy. Cancer Sci. 2008;99:1501–6.
Zhang Y, Daaka Y. PGE2 promotes angiogenesis through EP4 and PKA Cgamma pathway. Blood. 2011;118:5355–64.
Ninomiya I, Nagai N, Oyama K, et al. Antitumor and anti-metastatic effects of cyclooxygenase-2 inhibition by celecoxib on human colorectal carcinoma xenografts in nude mouse rectum. Oncol Rep. 2012;28:777–84.
Prosperi JR, Mallery SR, Kigerl KA, Erfurt AA, Robertson FM. Invasive and angiogenic phenotype of MCF-7 human breast tumor cells expressing human cyclooxygenase-2. Prostaglandins Other Lipid Mediat. 2004;73:249–64.
Nie D, Tang K, Szekeres K, Li L, Honn KV. Eicosanoid regulation of angiogenesis in human prostate carcinoma and its therapeutic implications. Ann N Y Acad Sci. 2000;905:165–76.
Nie D, Krishnamoorthy S, Jin R, et al. Mechanisms regulating tumor angiogenesis by 12-lipoxygenase in prostate cancer cells. J Biol Chem. 2006;281:18601–9.
Chatterjee M, Das S, Roy K, Chatterjee M. Overexpression of 5-lipoxygenase and its relation with cell proliferation and angiogenesis in 7,12-dimethylbenz(alpha)anthracene-induced rat mammary carcinogenesis. Mol Carcinog. 2013;52:359–69.
Sexton KR, Franzini L, Day RS, Brewster A, Vernon SW, Bondy ML. A review of body size and breast cancer risk in Hispanic and African American women. Cancer. 2011;117:5271–81.
Hyatt TC, Phadke RP, Hunter GR, Bush NC, Munoz AJ, Gower BA. Insulin sensitivity in African-American and white women: association with inflammation. Obesity (Silver Spring). 2009;17:276–82.
Colditz GA, Willett WC, Stampfer MJ, et al. Weight as a risk factor for clinical diabetes in women. Am J Epidemiol. 1990;132:501–13.
Autier P, Koechlin A, Boniol M, et al. Serum insulin and C-peptide concentration and breast cancer: a meta-analysis. Cancer Causes Control. 2013;24:873–83.
Rose DP, Vona-Davis L. The cellular and molecular mechanisms by which insulin influences breast cancer risk and progression. Endocr Relat Cancer. 2012;19:R225–41.
Blackburn GL, Wang KA. Dietary fat reduction and breast cancer outcome: results from the Women’s Intervention Nutrition Study (WINS). Am J Clin Nutr. 2007;86:s878–81.
Financial Disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Vona-Davis, L., Rose, D.P. The Obesity-Inflammation-Eicosanoid Axis in Breast Cancer. J Mammary Gland Biol Neoplasia 18, 291–307 (2013). https://doi.org/10.1007/s10911-013-9299-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10911-013-9299-z