
Abstract
This work reports the synthesis of two novel polyhedral oligomeric silsesquioxane (POSS) hybrid particles by investigating the effects of different crosslinking agents on morphology, porosity, chemical structure, crystalline properties, and thermal behavior of the resultant products. The hydrophilic glycerol dimethacrylate and hydrophobic ethylene glycol dimethacrylate (EGDMA) were used as co-monomer and crosslinking agents to obtain novel poly(POSS-co-GDMA) and poly(POSS-co-EGDMA) hybrid spherical microparticles by step-wise Pickering-like seeded polymerization. The crosslinking agent played a key role in specific surface area (SSA), average pore size, and pore volume, characterized by Brunauer–Emmett–Teller and Barrett–Joyner–Halenda analysis. When poly(POSS-co-GDMA) possess 88.0m2/g SSA, poly(POSS-co-EGDMA) has 3.5m2/g. Both particles exhibit a homogenous spherical shape in the polydisperse form and hybrid organosilica structure defined by scanning electron microscope, energy-dispersive X-ray spectroscopy, Fourier-transform infrared spectroscopy. The hybrid particles showed an amorphous silica composite character with thermal resistance up to 420 °C, determined by X-ray diffraction and thermogravimetric analysis. The mesoporous hybrid POSS particles could have great potential for many advanced material applications.
Graphical Abstract

Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Polyhedral oligomeric silsesquioxane (POSS) has significantly impacted hybrid material research with the advantages of the inorganic silica cage core surrounded by organic groups as a single molecule. Due to this unique nature, POSS is a candidate molecule for many application areas as the smallest possible particle of silica with its nanoscopic size of around 1–3 nm [1,2,3]. The organic groups provide an unlimited source of functionality to this hybrid molecule that enables the integration of various properties. The chemical (solubility [4], reactivity [5], inert [6], catalytic [7]), physical (wettability, conductivity [8], porosity [9]), optical [10], thermal [11], mechanical [12] properties of the hybrid material could be altered depending on the difference in the organic groups of POSS molecule.
Porosity is one of the key features of hybrid POSS materials, which could be classified according to their pore size as macro-, meso- and microporous, > 50 nm, 50–2 nm, and < 2 nm, respectively [13]. Particularly mesoporous POSS-based materials offer great promise in many applications such as separation technologies [14], drug delivery [15], sensor technologies [16], advanced coatings [17], and hydrogen storage [18]. The porosity of POSS-based materials was obtained via different methodologies, such as physical blending [19], chemical crosslinking [20], and grafting [21] using various POSS molecules. Among these methodologies, chemical crosslinking involves many reactions and techniques to obtain porous POSS-based materials. For example, Zhang et al. reported the different amounts of octavinylsilsesquioxane (OVS) and octahydrosilsesquioxane copolymerization in hydrosilation reaction to control specific surface areas from 380 to 530 m2/g via chemical crosslinking [22]. Chaikittisilp et al. synthesized hyper-crosslinked siloxane-organic hybrid material using benzyl-chloride-terminated cubic siloxane cage self-condensation via Friedel–Crafts alkylation and reached the highest BET surface area ∼2500 m2/g in siloxane-base materials [23]. Another POSS-based network was obtained through Schiff base chemistry. Guo et al. prepared the porous POSS-based polyimide functional aerogel through crosslinking with octa(aminophenyl)silsesquioxane that has 230 to 280m2/g using CO2 supercritical fluid extraction [24]. Alves et al. prepared vinyl-POSS monolith via thermally initiated free radical polymerization, resulting in a high BET surface area from 716 to 813m2/g [25].
Another vinyl substituent POSS monomer, Methacrly-POSS (M-POSS), was used for copolymerization reactions. In the study of Qu et al., M-POSS thermally polymerized in the presence of porogenic solvents to synthesize POSS-based hybrid monoliths via a free radical mechanism [26]. For this purpose, Qu et al. used two different hydrophobic crosslinking agents: bisphenol A dimethacrylate (BPADMA) and ethylene dimethacrylate (EDMA) 1:1 wt% ratio with M-POSS. While poly(POSS-co-BPADMA) monolith had 10.6m [2]/g BET surface area, poly(POSS-co-EDMA) monolith had 13.7m2/g with micropores > 1 µm in diameter [26].
Besides the monolith or bulk hybrid structures, the microparticle form POSS-based porous materials have recently been reported [27,28,29,30,31]. Bai et al. used various molar ratios up to 5% M-POSS as a co-monomer to synthesize EGDMA-based microparticles via the RAFT precipitation polymerization technique [27]. They reported that adding a little amount of hybrid M-POSS to the polymerization mixture increased the microparticle surface area from 20 to 78 m2/g [27]. A recent study by Bai et al. (2020) reported that 0.0085 wt% M-POSS/EGDMA microparticles were synthesized via RAFT precipitation polymerization, having ∼32m2/g BET surface area [28].
Our previous studies reported the synthesis of M-POSS hybrid microparticles via dispersion polymerization in a microfluidic reactor and emulsion polymerization in a batch-wise system [32, 33]. In the first study, while M-POSS microparticles were obtained in a microfluidic reactor, batch-wise synthesis resulted in a nanocluster form of M-POSS using one-step dispersion polymerization [32]. The second study reported micro/nano M-POSS particles via one-step emulsion polymerization with different surfactant combinations and ultrasound effects [33]. Although most M-POSS particles were around 300 nm, some micron-size poly(M-POSS) particles were obtained. However, the micron-size M-POSS particles were decorated by nano-size poly(M-POSS) due to colloidal instability explained by Ostwald ripening [33, 34].
This study aims to investigate the effects of different crosslinking agents on M-POSS particles using seeded emulsion polymerization. For this purpose, we added the same amount of hydrophilic GDMA and hydrophobic EGDMA to M-POSS particles. We observed that the seeded emulsion polymerization could help the diffusion mechanism of M-POSS and the stabilization of emulsion overcoming Ostwald ripening on M-POSS-based particles in an emulsion medium. The poly(POSS-co-GDMA) and poly(POSS-co-EGDMA) particles showed almost the same hybrid chemical structure, size distribution, crystalline structure, and thermal degradation behavior with different porosity characters defined by Fourier-transform infrared spectroscopy (FTIR), energy-dispersive X-ray spectroscopy (EDAX), scanning electron microscope (SEM), X-ray diffraction (XRD), and thermogravimetric analysis (TGA) and Brunauer–Emmett–Teller (BET) analysis.
2 Materials and Methods
2.1 Materials
The hybrid monomer, methacryl‐Polyhedral Oligomeric Silsesquioxane (M‐POSS) cage mixture (MA‐0735), was purchased from Hybrid Plastics Inc. The crosslinking agents, glycerol dimethacrylate (GDMA)-ethylene glycol dimethacrylate (EGDMA), and the template monomer glycidyl methacrylate were purchased from Sigma-Aldrıch. An emulsifying agent, sodium dodecyl sulfate (SDS), and stabilizers polyvinyl alcohol (PVA, 87–89% hydrolyzed, Mw = 85,000–146,000) and polyvinylpyrrolidone K30 (PVP‐K30) were obtained from Sigma‐Aldrich. The thermal initiator 2,2′‐Azobisizobutyronitrile (AIBN) and benzoyl peroxide (BPO) were also bought from Sigma‐Aldrich, which were recrystallized from methanol before use. Solvents ethylbenzene (EB), ethanol (EtOH), tetrahydrofuran (THF), and acetone were obtained from Sigma-Aldrich. Deionized water (dH2O) used in all experiments was obtained using Millipore/Direct Q-3UV.
2.2 Synthesis of Porous POSS Hybrid Particles
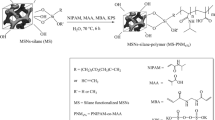
The poly(POSS-co-GDMA) and poly(POSS-co-EGDMA) particles were synthesized by seed emulsion polymerization. The polymerization occurred in two steps in Fig. 1. The seed particle poly(GMA) was synthesized in 2 µm size via dispersion polymerization [35]. Basically, 3 ml of GMA, 0.45 g of PVP-K30, and 0.24 g of AIBN were dissolved in 30 ml of EtOH by 5 min ultrasonication. The polymerization was carried out at 70 °C overnight. Obtained poly(GMA) particles were washed with EtOH and water.

Schematically representation of A two-step polymerization and B chemical structures of the hybrid monomer methacryl-POSS, two different crosslinking agents hydrophilic structure GDMA with hydroxyl (–OH) group and hydrophobic structure EGDMA with methyl (–CH3)
The template particle poly(GMA) was swollen by EB [36, 37]. A certain amount of poly(GMA) was added to SDS containing medium in Table 1. The medium was emulsified by an ultrasonic probe (Qsonica, Q500 sonicator) in the ice‐cooled bath for 5 min. The suspension was magnetically stirred overnight at room temperature.
The organic phase was prepared by dissolving the main monomer M-POSS, different crosslinking agents (GDMA and EGDMA), and the initiator (BPO) in acetone. The amounts of polymerization composition are given in Table 1. The mixture was added into SDS containing emulsified medium. This emulsion was added into step 1 suspension and magnetically stirred overnight. Before polymerization, the 8% (w/v) PVA solution was added to the final polymerization medium. The polymerization was carried out at 80 °C for 24 h under moderately magnetic stirring. After polymerization, the particles were collected by centrifuge from the polymerization medium. The unreacted medium was removed from particles. The obtained particles were washed water, EtOH, THF, EtOH, and water. The first washing step was to remove the surfactant from the collected particles by water. EtOH removed the unreacted monomers. The seed poly(GMA) particles were dissolved and removed by THF and EtOH. The remaining particles were kept either in solvent (water, EtOH) or dried as white powder form.
2.3 Morphological Characterization
The morphological properties of porous poly(POSS-co-GDMA) and poly(POSS-co-EGDMA) particles were characterized by scanning electron microscopy SEM (Quanta 450 Akishima, Tokyo, Japan). For the sample preparation, the particle solution was mixed via vortex, and the 2 µl of particle solution was dispersed onto double‐sided sticky carbon tapes fixed to a flat sample holder and kept for 5 min to air‐dry. The samples were coated with 6 nm Au–Pt thin-film to obtain a conductive surface and protect the particles from thermal degradation during SEM analysis.
2.4 Chemical Structure Analysis
The surface chemistry of the particles was analyzed by energy-dispersive X-ray spectroscopy EDAX (Quanta 450 Akishima, Tokyo, Japan). The organic and inorganic chemical bond structures of monomer and particle were determined by Fourier‐transform infrared spectroscopy FTIR (Thermo Scientific Nicolet™, USA). FTIR spectrums were collected for the powder form of particles and liquid form of monomers by scanning between 4000 and 500 cm−1.
2.5 Porosity and Pore Size Distribution Analysis
Specific surface areas, pore sizes, and pore volumes of the particles were determined by conventional Brunauer–Emmett–Teller (BET) analysis nitrogen adsorption measurements using Quantachrome Nova 2200e series volumetric gas adsorption instrument. The samples were prepared as pre-dried at 50 °C overnight and weighed 0.3 g by each synthesized particle. The pre-dried particles were put into a BET cell and left for 6 h at degassing station at 80 °C in the BET analyzer. The cells were taken to the analysis part, and approximately half a litter of nitrogen was used for each analysis. The experiments for adsorption isotherms were performed at -195.85 °C using liquid nitrogen.
2.6 Thermal Degradation Analysis
The thermal decomposition temperature of the particles was measured using a thermogravimetric analyzer (Hitachi STA7300, Japan) in a nitrogen atmosphere with a heating rate of 10 °C/min ranging from 25 to 500 °C. Approximately 10 mg of sample was put into aluminum TGA crucibles for the analysis. The degradation steps were determined at the transition points where the curve passed the maximum negative slope.
2.7 Crystalline Structure Analysis
The crystalline structure of the particles was determined by X-Ray diffraction and XRD (Rigaku Miniflex 600 X-Ray diffractometer, Japan). The powder form of the samples was analyzed at 40 kV, 15 mA with 2.000 deg/min of scanning speed, 0.0200 deg of step width, and from 5° to 90° scanning range.
3 Results and Discussion
3.1 Morphological and Chemical Structure
The POSS-based mesoporous microparticles were obtained by two-seep seed polymerization. The morphological structure was investigated by SEM in Fig. 2. The resultant product, poly(POSS-co-GDMA) and poly(POSS-co-EGDMA) microparticles, have a spherical form. Although both particles possess similar shapes, their pore structure and surface morphology differ. While the porous structure could be easily seen in poly(POSS-co-GDMA) particles (Fig. 2A), poly(POSS-co-EGDMA) particles exhibited a non-porous structure in SEM images (Fig. 2B).

SEM images and EDX spectrums of synthesized particles A poly(POSS-coGDMA) data, B poly(POSS-co-EGDMA) with ×50,000 and ×100,000 magnification respectively and scale bar: 1 µm
The particle size distribution was polydisperse for both hybrid particles in Figure S1. The particle size of poly(POSS-co-GDMA) varies between 0.69 and 5.61 µm with 2.08 µm \(\pm\) 1.24 average size in Fig. S1-A. The particles size of poly(POSS-co-EGDMA) changes from 0.55 to 2.00 µm with 1.00 µm \(\pm\) 0.28 average size in Figure S1-B. While hydrophobic crosslinking agent (EGDMA) resulted in a more narrow distribution with a non-porous structure, hydrophilic one (GDMA) had a broad size distribution with a porous structure. Even though GDMA and EGDMA have the same chain length (Fig. 1B), the hydrophilic character of hydroxyl –OH carrying GDMA affected the polymerization resulting in size distribution and porosity. The solubility in the reaction medium and diluent differs due to the hydroxyl (–OH) functionality of GDMA. Hence, hydrophilic GDMA should be slightly more polar, have a higher solubility in an aqueous medium, and could diffuse in swollen seed poly(GMA) particles. That could also explain the bigger particle size in poly(POSS-co-GDMA) in Fig. S1.
The common acceptance of the multi-stage seed polymerization mechanism is based on diffusion and agglomeration of the monomer phase within the seed particles [36, 38, 39]. When the solubility of the monomer is high in the diluent, the monomer mixture diffuses into seed particles. The higher polarity difference between the monomer phase and the diluent within the swollen particles resulted in faster phase separation, forming crosslinked nuclei and excessive aggregation to generate solid particles. In this study, M-POSS monomer and crosslinking agents (GDMA and EGDMA) dissolved in acetone to form the monomer phase. According to the multi-stage seed polymerization approach, the monomer phase should diffuse in linear poly(GMA) seed particles swollen by ethylbenzene, resulting in high monodispersity. However, we understand that the diffusion mechanism, similar to methacrylate monomers, did not work as it was accepted (Fig. 1). We hypothesize that even though the M-POSS monomer carries a methacryl- functional group, diffusion could not efficiently occur into seed particles due to the high molecular weight and hybrid structure of the M-POSS monomer. Besides the diffusion mechanism through seed particles, homogeneous coagulative nucleation of the monomer phase could create reaction loci in the emulsion medium to form sub-micron particles. Therefore, the swollen poly (GMA) seed particle became a template and a solid stabilizer for the polymerization of hybrid M-POSS, similar to Pickering emulsion polymerization [40, 41].
Both poly(POSS-co-GDMA) and poly(POSS-co-EGDMA) particles exhibit organic and inorganic hybrid structures with Carbon (C), Silicon (Si), and Oxygen (O) content in Fig. 2. The chemical structure and functional groups were analyzed using FTIR analysis in Fig. 3 and Table 2. Here, the POSS-based monomer and crosslinking agents (GDMA and EGDMA) carried the vinyl group (–C=C–) for thermally initiated-free radical polymerization [33, 42]. After the complete reaction, the peak of the vinyl functional group (from 1635 to 1660 cm−1) diminished at the final product poly(POSS-co-GDMA) and poly(POSS-co-EGDMA) particles [33]. The organic and inorganic structure (–Si–O–Si– peak at 1100 cm−1) of the POSS-based monomer was observed in both particles [42]. The only functional group difference between poly(POSS-co-GDMA) and poly(POSS-co-EGDMA) is the hydroxyl functional group that entered the structure via GDMA. The hydrophilic character of GDMA (–OH board peak at 3500 cm−1) was detected in hybrid poly(POSS-co-GDMA) particles [42]. The other peaks at 815 cm−1, 835 cm−1, and 740 cm−1 represent (CH3)3–Si–O symmetric stretch related to the POSS part of both hybrid particles in the FTIR spectrum [43].

FTIR spectrum of crosslinking agents, main monomer, and synthesized particles (a) GDMA, (b) EGDMA, (c) POSS, (d) poly(POSS-co-GDMA), and (e) poly(POSS-co-EGDMA)
3.2 Porosity and Pore Structures
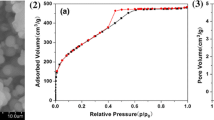
The specific surface area (SSA) and pore size distribution of poly(POSS-co-GDMA) and poly(POSS-co-EGDMA) were evaluated using BET and BJH methods. The porosity properties are given in Table 3. While poly(POSS-co-GDMA) particles have 88.0m2/g SSA with mesopore structure, the SSA of poly(POSS-co-EGDMA) particles find 3.5m2/g using BET analysis in Fig. S2. Poly(POSS-co-GDMA) has higher pore SA and pore volume (126m2/g and 0.188 cm3/g, respectively) than poly(POSS-co-EGDMA), which has 1.5m2/g and 0.008 cm3/g. The SSA, pore SA, and Pore volume results were consistent with SEM photos, as poly(POSS-co-GDMA) particles have a porous structure.
The pore size distribution was found by cumulative pore volume (cm3/g) dependent on the pore diameter (nm) in Fig. 4. Here poly(POSS-co-GDMA) showed a bi-modal pore diameter of 3.7 nm and 6 nm with mesoporous character. Even though the pore diameter of poly(POSS-co-EGDMA) was calculated at approximately 4 nm using BJH Method, the SEM images and the pore volume to pore diameter ratio showed the non-porous character of the particle. During porosity analysis, the pore space formed between the polydisperse poly(POSS-co-EGDMA) particles that create the porosity properties of them in Table 3.

Nitrogen adsorption analysis for pore volume distribution and differential pore volume distribution of A poly(POSS-co-GDMA), B poly(POSS-co-EGDMA)
The crosslinking density of the polymeric network directly affected the porosity [36, 44]. According to the proposed polymerization mechanism, phase separation formed the crosslinked nucleic due to the polarity difference between the monomer solution and diluent [36, 40]. When the polarity difference increases, phase separation occurs faster, forming large pores and aggregates. In the contrary case, the similar polarity between the monomer and diluent results in slower phase separation, generating smaller pores [36]. While polarity is one important parameter to obtain porous structures in Pickering-like seeded polymerization, the H-bonding ratio, the total solubility ratio, and the viscosity ratio affected the porosity of the particles, too [45]. As we mentioned, hydroxyl (-OH) carrying GDMA is more hydrophilic than EGDMA. Therefore, the polarity difference between GDMA and diluents (hydrophobic character non-polar compound ethylbenzene) leads to faster phase separation, resulting in a porous structure. On the contrary, the polarity of the hydrophobic crosslinking agent EGDMA is closer to the diluents and methacryl- carrying monomer that resulted with smaller fixed aggragates with non-porous strucutre.
3.3 Crystalline Structure
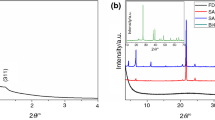
X-ray diffraction (XRD) evaluated both hybrid particles’ crystalline structure, and the diffractograms are shown in Fig. 5. The XRD peaks of poly(POSS-co-GDMA) and poly(POSS-co-EGDMA) particles had the same amorphous characteristics with a board peak at 17°. Similar board peak characteristics were reported at 2θ = 17.12 for equivalent hexagonal cells [46].

XRD pattern of (a) poly(POSS-co-GDMA), (b) poly(POSS-co-EGDMA)
The XRD pattern confirmed the polymerization along with the formation of amorphous silica. The crystalline silica structure could only be seen with sharp peaks along with the board peak. In this case, no sharp peak was present in the diffractogram. Therefore, both particles do not carry a crystalline silica structure. However, the cage structure of POSS in polymeric particles provided a hexagonal lattice form peak.
3.4 Thermal Properties
The thermal stability of POSS-hybrid particles was determined using thermogravimetric analysis (TGA). The percentage mass loss and derivative thermogravimetry (DTG) curves of particles are in Fig. 6. The TG and DTG curves of poly(POSS-co-GDMA) were represented by a black straight line and a black dash line, respectively. The red color dot line and dash-dot line described TG and DTG of poly(POSS-co-EGDMA), respectively. The significant steps of thermal decomposition were given as inflection points and percentage decomposition (ΔY%) in Table 4.

TG (mass %) and DTG (µg/min) of (a) poly(POSS-co-GDMA), (b) poly(POSS-co-EGDMA)
The first step represents the residual acetone in the hybrid particle structure as approximately 2% of the total mass. The remaining hybrid particles exhibited slightly different thermal degradation profiles (Fig. 6). 6.5% carbon content (volatile alkyl chains) of Poly(POSS-co-EGDMA) particles degraded at 268 °C. In the next degradation step, the remaining organic content left both particle structures at 330 °C. The main degradation step was observed around 420 °C as a sharp peak representing the cleavage of C–C and Si–C bonds [47]. The slow and weak peak around 490 °C showed the transition into the ceramic for poly(POSS-co-GDMA) particles. 80% of the hybrid material decomposed till 550 °C.
Many studies support the flame-retardant property of POSS molecules forming a char layer that protects the bulk materials from heat transfer. POSS-based hybrid molecules are used in several polymers as flame retardant additives to increase thermal stability, such as polyurethane (PU) [46], epoxy [48], and polyethylene terephthalate (PET) [49]. Poly(POSS-co-GDMA) and poly(POSS-co-EGDMA) could be promising additives or coating materials that exhibit high thermal resistance.
4 Conclusion
Mesoporous organosilica materials have always been attractive to researchers due to the variety of application areas [50]. Porous or non-porous silica-based organic composite particles are commonly obtained using silica precursors with well-known Stöber or sol–gel processes. In this study, an alternative to common silica precursors and organosilica synthesis processes, the M-POSS hybrid molecule polymerized with different crosslinking agents (GDMA and EGDMA). Even though the polymerization conditions and chain lengths of the crosslinking agent are the same, the resultant products poly(POSS-co-GDMA) exhibited mesoporous characteristics with 88m2/g SSA, poly(POSS-co-EGDMA) had nonporous character. Both particles showed organic and inorganic hybrid structures in a uniform spherical shape with excellent heat resistance properties up to 420 °C. The hybrid particles have great potential for multipurpose applications such as chromatographic separations, absorbents, catalyst carriers, composite additives, and thermal insulation.
References
S.A. Madbouly, A. Lendlein, Shape-memory polymer composites, in Shape-memory polymers. (Springer, Berlin, 2009), pp.41–95
J.D. Lichtenhan, K. Pielichowski, I. Blanco, Polymers 11, 1727 (2019)
H. Shi, J. Yang, M. You, Z. Li, C. He, Polyhedral oligomeric silsesquioxanes (POSS)-based hybrid soft gels: molecular design, material advantages, and emerging applications. ACS Mater. Lett. 2, 296–316 (2020)
H.W. Milliman, D. Boris, D.A. Schiraldi, Experimental determination of Hansen solubility parameters for select POSS and polymer compounds as a guide to POSS–polymer interaction potentials. Macromolecules 45, 1931–1936 (2012)
S. Zhang, Q. Zou, L. Wu, Preparation and characterization of polyurethane hybrids from reactive polyhedral oligomeric silsesquioxanes. Macromol. Mater. Eng. 291, 895–901 (2006)
X. Wang et al., Fluorinated polyhedral oligomeric silsesquioxanes. RSC Adv. 5, 4547–4553 (2015)
G. Kibar, D.ŞÖ. Dinç, In-situ growth of Ag on mussel-inspired polydopamine@ poly (M-POSS) hybrid nanoparticles and their catalytic activity. J. Environ. Chem. Eng. 7, 103435 (2019)
P. Zhang et al., Polydopamine-modified sulfonated polyhedral oligomeric silsesquioxane: an appealing nanofiller to address the trade-off between conductivity and stabilities for proton exchange membrane. J. Membr. Sci. 596, 117734 (2020)
M. Ahmed, H. Ghanbari, B.G. Cousins, G. Hamilton, A.M. Seifalian, Small calibre polyhedral oligomeric silsesquioxane nanocomposite cardiovascular grafts: influence of porosity on the structure, haemocompatibility and mechanical properties. Acta Biomater. 7, 3857–3867 (2011)
H. Tunstall-Garcia, B.L. Charles, R.C. Evans, The role of polyhedral oligomeric silsesquioxanes in optical applications. Adv. Photo. Res. (2021). https://doi.org/10.1002/adpr.202000196
M.G. Mohamed et al., Ultrastable porous organic/inorganic polymers based on polyhedral oligomeric silsesquioxane (POSS) hybrids exhibiting high performance for thermal property and energy storage. Microporous Mesoporous Mater. 328, 111505 (2021)
K. Mishra, G. Pandey, R.P. Singh, Enhancing the mechanical properties of an epoxy resin using polyhedral oligomeric silsesquioxane (POSS) as nano-reinforcement. Polym. Test. 62, 210–218 (2017)
M.T. Gokmen, F.E. Du Prez, Porous polymer particles—A comprehensive guide to synthesis, characterization, functionalization and applications. Prog. Polym. Sci. 37, 365–405 (2012)
H.-B. He et al., Fabrication of enrofloxacin imprinted organic–inorganic hybrid mesoporous sorbent from nanomagnetic polyhedral oligomeric silsesquioxanes for the selective extraction of fluoroquinolones in milk samples. J. Chromatogr. A 1361, 23–33 (2014)
S. Gandhi et al., Synthesis of a novel hierarchical mesoporous organic–inorganic nanohybrid using polyhedral oligomericsilsesquioxane bricks. New J. Chem. 38, 2766–2769 (2014)
M. Seino et al., Low-k periodic mesoporous organosilica with air walls: POSS-PMO. J. Am. Chem. Soc. 133, 18082–18085 (2011)
Y. Yin et al., “Open-mouth” mesoporous hollow micro/nano coatings based on POSS/PDMS: fabrication, mechanisms, and anti-icing performance. Part. Part. Syst. Charact. 35, 1800323 (2018)
Z. Wei, X. Luo, L. Zhang, M. Luo, POSS-based hybrid porous materials with exceptional hydrogen uptake at low pressure. Microporous Mesoporous Mater. 193, 35–39 (2014)
S. Bandehali, F. Parvizian, A. Moghadassi, S.M. Hosseini, High water permeable PEI nanofiltration membrane modified by L-cysteine functionalized POSS nanoparticles with promoted antifouling/separation performance. Sep. Purif. Technol. 237, 116361 (2020)
X. Yang et al., The preparation and chemical structure analysis of novel POSS-based porous materials. Materials 12, 1954 (2019)
L. Hao et al., Low dielectric and high performance of epoxy polymer via grafting POSS dangling chains. Eur. Polym. J. (2022). https://doi.org/10.1016/j.eurpolymj.2022.111313
C. Zhang et al., Highly porous polyhedral silsesquioxane polymers. Synthesis and characterization. J. Am. Chem. Soc. 120, 8380–8391 (1998)
W. Chaikittisilp et al., Porous siloxane–organic hybrid with ultrahigh surface area through simultaneous polymerization–destruction of functionalized cubic siloxane cages. J. Am. Chem. Soc. 133, 13832–13835 (2011)
H. Guo et al., Polyimide aerogels cross-linked through amine functionalized polyoligomeric silsesquioxane. ACS Appl. Mater. Interfaces 3, 546–552 (2011)
F. Alves, P. Scholder, I. Nischang, Conceptual design of large surface area porous polymeric hybrid media based on polyhedral oligomeric silsesquioxane precursors: preparation, tailoring of porous properties, and internal surface functionalization. ACS Appl. Mater. Interfaces 5, 2517–2526 (2013)
J. Ou, Z. Zhang, H. Lin, J. Dong, H. Zou, Polyhedral oligomeric silsesquioxanes as functional monomer to prepare hybrid monolithic columns for capillary electrochromatography and capillary liquid chromatography. Anal. Chim. Acta 761, 209–216 (2013)
J. Bai et al., Synthesis and characterization of paclitaxel-imprinted microparticles for controlled release of an anticancer drug. Mater. Sci. Eng. C 92, 338–348 (2018)
J. Bai et al., Synthesis and characterization of molecularly imprinted polymer microspheres functionalized with POSS. Appl. Surf. Sci. 511, 145506 (2020)
Y.-Y. Deng et al., Monodispersed hybrid microparticles based on polyhedral oligomeric silsesquioxane with good UV resistance and high thermal stability: from organic to inorganic. Polymer 178, 121609 (2019)
D. Han et al., Facile construction of porous magnetic nanoparticles from ferrocene-functionalized polyhedral oligomeric silsesquioxane-containing microparticles for dye adsorption. Ind. Eng. Chem. Res. 59, 9532–9540 (2020)
D. Han et al., Engineering the surface pattern of microparticles: from raspberry-like to golf ball-like. ACS Appl. Mater. Interfaces. 13, 31215–31225 (2021)
G. Kibar, U. Çalışkan, E.Y. Erdem, B. Çetin, One-pot synthesis of organic–inorganic hybrid polyhedral oligomeric silsesquioxane microparticles in a double-zone temperature controlled microfluidic reactor. J. Polym. Sci. Part A 57, 1396–1403 (2019)
G. Kibar, Spherical shape poly (M-POSS) micro/nano hybrid latex particles: one-step synthesis and characterization. J. Appl. Polym. Sci. 137, 49241 (2020)
K.-J. Kim, Nano/micro spherical poly (methyl methacrylate) particle formation by cooling from polymer solution. Powder Technol. 154, 156–163 (2005)
B. Elmas, M. Tuncel, G. Yalçın, S. Şenel, A. Tuncel, Synthesis of uniform, fluorescent poly (glycidyl methacrylate) based particles and their characterization by confocal laser scanning microscopy. Colloids Surf. Physicochem. Eng. Aspects 269, 125–134 (2005)
G. Kibar, A. Tuncel, Synthesis and characterization of monodisperse-porous, zwitterionic microbeads. Polym. Bull. 73, 1939–1950 (2016)
G. Kibar, A. Tuncel, Gold-nanoparticle decorated monosized magnetic polymer based catalyst: reduction of 4-nitrophenol. J. Inorg. Organomet. Polym Mater. 28, 2249–2257 (2018)
A. Tuncel, M. Tuncel, B. Ergun, C. Alagöz, T. Bahar, Carboxyl carrying-large uniform latex particles. Colloids Surf. A 197, 79–94 (2002)
C. Cheng, J. Vanderhoff, M. El-Aasser, Monodisperse porous polymer particles: formation of the porous structure. J. Polym. Sci. Part A 30, 245–256 (1992)
H. Ma, M. Luo, S. Sanyal, K. Rege, L.L. Dai, The one-step pickering emulsion polymerization route for synthesizing organic-inorganic nanocomposite particles. Materials 3, 1186–1202 (2010)
H. Jiang, Y. Sheng, T. Ngai, Pickering emulsions: versatility of colloidal particles and recent applications. Curr. Opin. Colloid Interface Sci. 49, 1–15 (2020)
Y. Liu, Y. Huang, L. Liu, Influences of monosilanolisobutyl-poss on thermal stability of polymethylsilxoane. J. Mater. Sci. 42, 5544–5550 (2007)
O. Toepfer, D. Neumann, N.R. Choudhury, A. Whittaker, J. Matisons, Organic–inorganic poly (methyl methacrylate) hybrids with confined polyhedral oligosilsesquioxane macromonomers. Chem. Mater. 17, 1027–1035 (2005)
M. Sobiesiak, Analysis of structure and properties of DVB–GMA based porous polymers. Adsorption 25, 257–266 (2019)
F.R. Mansour, S. Waheed, B. Paull, F. Maya, Porogens and porogen selection in the preparation of porous polymer monoliths. J. Sep. Sci. 43, 56–69 (2020)
B.X. Fu et al., Nanoscale reinforcement of polyhedral oligomeric silsesquioxane (POSS) in polyurethane elastomer. Polym. Int. 49, 437–440 (2000)
Y. Liu, Y. Huang, L. Liu, Thermal stability of POSS/methylsilicone nanocomposites. Compos. Sci. Technol. 67, 2864–2876 (2007)
G. Kibar, Epoxy Functional Porous POSS Microparticle Synthesis. Hacett. J. Biol. Chem. 50, 359–366 (2022)
H. Sirin, D. Turan, G. Ozkoc, S. Gurdag, POSS reinforced PET based composite fibers:“effect of POSS type and loading level.” Compos. B Eng. 53, 395–403 (2013)
J.G. Croissant, Y. Fatieiev, A. Almalik, N.M. Khashab, Mesoporous silica and organosilica nanoparticles: physical chemistry, biosafety, delivery strategies, and biomedical applications. Adv. Healthcare Mater. 7, 1700831 (2018)
Acknowledgements
Thanks to the Turkish Fulbright Commission for a Fulbright Postdoctoral Scholarship and The Scientific and Technological Research Council of Turkey (TUBITAK 2219 Postdoctoral Award #1059B191801017) for Dr. Gunes Kibar.
Funding
This work was supported by Turkish Fulbright Commission, Fulbright Postdoctoral Scholarship, The Scientific and Technological Research Council of Turkey, TUBITAK 2219 Postdoctoral Award #1059B191801017
Author information
Authors and Affiliations
Contributions
Author Statement Gunes Kibar: Conceptualization, Investigation, Formal analysis, Resources, Project administration, Writing - Original Draft
Corresponding author
Ethics declarations
Conflict of interest
The author declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kibar, G. Effect of Crosslinking Agent on Mesoporous Spherical POSS Hybrid Particles: Synthesis, Characterization and Thermal Stability. J Inorg Organomet Polym 33, 831–840 (2023). https://doi.org/10.1007/s10904-023-02540-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10904-023-02540-z