
Abstract
Nanocrystalline Pd@PdO core–shell (a) were synthesized by aqueous sol–gel transformations from precursor [Cl2PdL] (1) {where, L = 8-(2-pyridylmethoxy)quinoline}. Formation of the precursor (1) was confirmed by elemental analysis, molecular weight measurements, FT-IR, and NMR (1H and 13C). PdO nanoparticles (b) were obtained when unmodified PdCl2 (2) was used as precursor. XRD studies were done to determine crystal structure and phases of synthesized powders which was found to be cubic, tetragonal (Pd@PdO) and tetragonal (PdO), respectively. TEM studies were done to confirm the results obtained from XRD. HRTEM and SAED patterns confirmed the formation of core–shell nanoparticles. Crystallite size of both the samples as approximated by XRD and TEM were found to be in nanometer range. Raman spectroscopy confirmed the presence of PdO in both the samples. Surface morphologies of both the samples were evaluated by SEM. The absorption spectra of samples, (a) and (b) show the energy band gap of 1.43 and 1.89 eV, respectively.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Palladium is a well-known rare metal which has generally been used in catalytic applications [1]. The nanoparticles of palladium are currently under intense research due to their enhanced performance in catalysis, hydrogen generation, electrochemical reactions and sensing applications [2,3,4,5]. Palladium is highly reactive thus forms oxide immediately. A lot of work has been done in the synthesis of palladium oxide (PdO) and studying them in various applications [6,7,8]. However, PdO varies in structure and performance in various applications than Pd [9, 10]. Core–shell Pd@PdO nanoparticles can be a new potential material in this regard in which core is of Pd and shell is of PdO. This allows a pristine Pd covered under oxide of the same which can be used for better performance. Various traditional methods have been explored for the synthesis of nanosized Pd including heat decomposition [11], oil-bath heating [12], Microwave irradiation [13] and biosynthesis methods [14]. All these methods led to synthesis of PdO rather than Pd. The synthesis of PdO was generally prepared by heat decomposition of Pd(NO3)2 was required a longer heating time at a higher temperature, the oil-bath heating produces significantly agglomerated particles while the microwave approach requires special chemicals and polymers and biosynthesis may lead to trouble in purification and influence properties of the target materials [15]. The successful attempts to synthesize the Pd@PdO nanoparticles were by Jose and Jagirdar [16] via solventless thermolysis method, Huang et al. [17] via reactive sputtering, and Jiang et al. [18] via chemical reduction method involving nitrogen doped graphene support.
Currently, the synthesis of nanostructures material via sol gel method is attracting great interest due to the obvious advantages, such as economics, energy efficiency, and environmental friendliness [19,20,21,22]. It also has a capability to control crystal structure as well as particle’s morphology by changing different parameters like precursor electronic environment, concentration, pH, processing temperature, etc [22,23,24].
The natures of ligands, the coordination number of central metal and the rate of hydrolysis are also control the desired morphology and the size of nanostructures [25, 26]. Within the promising material chemistry, there is no report so far on the sol gel synthesis of PdO.
Hence in this paper, we report the synthesis of Pd@PdO core–shell nanoparticles by the sol–gel transformation of [PdCl2·(C15H12Cl2N2O)]. The generation of the core–shell Pd@PdO and general PdO were affected by the precursor chemistry used in the synthesis. PdO nanoparticles were obtained by sol–gel transformation of PdCl2.
2 Experimental
2.1 General Procedure and Materials
8-hydroxyquinoline (99%) was purchased from Sigma-Aldrich and purified by sublimation prior to use. Palladium (II) chloride was obtained from Thermo-fisher Scientific (India). The solvents used in the experimental work, were of analytical grade and purchased from Rankem, India, and dried by conventional methods and distilled prior to use [27]. All manipulations for the syntheses of precursor (1) were carried out in strictly anhydrous conditions. Ligands were synthesized as reported in the literature [28]. Palladium was estimated as PdO and nitrogen using Kjeldahl’s method [27]. IR spectra [4000–400 cm−1] were recorded as dry KBr pellets on a PerkinElmer Spectrum Version 10.4.00, FT-IR spectrometer. NMR [1H and13C{1H}] data were collected on JEOL FX 300 FT-NMR spectrometer in CDCl3 or DMSO-d6 solution at 400.4 MHz and 75.45 MHz frequencies for 1H and 13C{1H} NMR spectra, respectively. The molecular weight measurements were carried out by the elevation in boiling point method using a Beckmann thermometer fitted in a glass assembly (supplied by JSGW, India) in anhydrous benzene. Thermogravimetric analyses of palladium complex were performed on PerkinElmer Pyris. The measurements were done at a heating rate of 10 °C/min from 30 to 800 °C under flowing nitrogen environment. The XRD patterns of both the samples were recorded on P’analytical X’Pert PRO MPD diffractometer (model 3040). The X-ray were produced using a sealed tube and the wavelength of X-ray was 0.154 nm (Cu Kα). SEM and EDX was performed on Carl-Zeiss (30 keV) make and model EVO. The powder samples were used for SEM and EDX. TEM was carried out on TECHNAI 20, FEI, North America, and Bruker, Germany. The particles were dispersed in the methanol then a drop of dispersion was added on the carbon-coated copper grid. The copper grid was then left to dry overnight before the TEM studies. Raman spectra were performed using STR-500 confocal micro RAMAN spectrometer (Ramansystem, USA) having excitation laser of k = 532 nm. Raman spectra were recorded in the range 200–1500 cm−1. The C, H, N analyses were performed on Thermo scientific Flash 2000. Absorption spectra of all the samples (a) and (b) were recorded using UV–Vis spectrophotometer in 200 to 800 nm wavelength range. Tauc method was used to obtain the band gap of the synthesized nanoparticles.
2.2 Synthesis of Palladium Complexes
2.2.1 Synthesis of Palladium Complexes of 8-(2-Pyridylmethoxy)Quinoline or [PdCl2L]
An amount of 0.32 g (1.35 mmol) of PdCl2 was added to the MeCN solution of ligand 8-(2-pyridylmethoxy)quinoline (0.24 g, 1.35 mmol in ~ 20 ml of MeCN) in 1:1 molar ratio. The reaction mixture was stirred at room temperature for 5 min. The resulting mixture was filtered and washed with dichloromethane and dried under vacuum to give dark orange colored solid, which was used as the source material for synthesizing Pd@PdO. The physical, analytical and spectral data of the precursor are given below:
[PdCl2·(C15H12Cl2N2O)]:[yield: 0.43 g, 76%], mp. 231.4 °C, M.W. obs. (calc.): 402.87 (411.94); analyses (%) obs. (calc.): C: 43.55 (43.56), H: 2.93 (2.92), N: 6.73 (6.77),Cl: 17.13 (17.14),Pd: 25.71 (25.73); FT-IR, ν, cm−1: 2926 (C–H);1553 (C=N); 1613 (C = car, asym); 1468 (C = car, sym); 1386 (C–O–alkyl chain); 111]C-Oar). 575 (Pd–O), 423 (Pd–N), 1H NMR (400.13 MHz, DMSO-d6, at 25 °C, δ ppm): 8.11 (d, H, H2); 8.98 (d, H, H7);8.61 (d, H, H of C=N Py);7.64–7.05 (m, 7H, other aromatic protons); 5.56 (s, 2H, H of –O–CH2–). 13C NMR (DMSO-d6, 75.4 MHz): δ 157.1 (C-8); δ 153.9 (C-2); 149.4 (C-15); δ 140.1 (C-10), δ 137.2 (C-9), δ 137.2 (C-16), δ 71.4 (C-11), 29.2 (C-12), 25.9 (C-13), 129.6, 126.8, 122.8, 121.6, 120.2, 109.9 (Ar. Carbon).
2.2.2 Sol–Gel Transformation of [PdCl2L] (1) and PdCl2 (2) to Synthesize Pure Pd@PdO (a) and Palladium Oxide (PdO) (b)
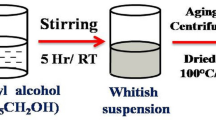
[PdCl2L] (1) (0.32 g, 0.77 mmol) was dissolved in ethanol (~ 20 ml) and THF. To this dirty yellow colored solution, 2 ml of ammonia solution was added, gelation occurs immediately and the colour turned to dark yellow. The solution was stirred for 24 h, some more quantity of ammonia solution (∼ 5 ml) was again added and the mixture was stirred for 2 days to ensure complete hydrolysis. The resulting gel was dried in a preheated oven (~ 100˚C),dark greenish colored solid was obtained, which was washed several times with deionized water and the mixture of n-hexane and acetone. The above solid was sintered at 600 °C for 3 h to give dark green colored powders, which was characterized as pure Pd@PdO.
PdCl2 (2) was also hydrolyzed by the above similar route and sintered at 600 °C to yield the dark green colored powder and characterized as PdO.
3 Results and Discussion
3.1 Synthesis and Characterization of Precursor (1)
A systematic study of the reaction of palladium dichloride with tridentates ligand (L) in 1:1 molar ratio in MeCN has been carried out and gives products of the type, [PdCl2L] in quantitative yield. The reaction can be represented by following equation:
The above mentioned reaction was found to be quite facile and quantitative. The complex is colored solid and insoluble in water partially soluble in ethanol and methanol but completely soluble in DMSO. The molecular weight determination of the newly synthesized complex indicated monomeric nature of the compound in solution phase.
3.1.1 IR Spectra
The important IR spectral bands and their assignments of precursor are presented in 2 section. These assignments were made by comparing the IR spectra with those of the ligand and its ligand complex [28]. IR spectra (Fig. 1, Supplementary Materials) of ligands show strong bands at 2990–3050 cm−1, attributable to the symmetric stretching of O–CH2 group vibrations. In the spectrum of the ligand, the appearance of ν(C=N) stretching vibration at 1640–1665 cm−1 is shifted to the lower-frequency region (1568–1616 cm−1) in the corresponding palladium complex, suggesting the coordination of the nitrogen atom to the palladium, which is further supported by the new bands appeared in the region (420–429 cm−1) assigned to v(Pd–N) in the above-mentioned complex [29]. The band appeared in the region 575 cm−1 may be due to the (Pd–O) vibrations, are support the coordination of oxygen to metal atom [30]. The medium intensity band observed in the region of 1256–1270 cm−1 is assigned to ν(C–O) of the alkoxy chain in IR spectra of all the above complexes. The study and comparison of infrared spectra of ligand and its metal complex imply that the ligand behave as mono basic tridentate ligand.

Sol–gel synthesis of Pd@PdO, and PdO nanoparticles
3.1.2 NMR Spectra
The 1H and 13C NMR spectra (Figs. 2, 3, Supplementary Materials) of ligand and its metal complex were recorded in DMSO-d6. The spectral data of the palladium complex was summarized in 2 section and interpreted by comparing with the spectra of parent organic ligand (L) and its reported metal complexes [28]. These comparisons indicate that –CH=N proton of the pyridine rings and proton of the phenoxy rings in ligand shifted to the downfield in the complex consistence with coordination of the nitrogen and oxygen. The other proton of the ligand did not show significant differences between ligands and its metal complexes and reveal characteristic signals at the expected position with desired multiplicity.

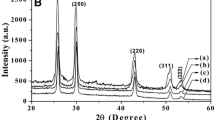
Powder XRD patterns of: a Pd@PdO and b PdO

a TEM image of sample (a) with particle size distribution in inset; b TEM image of sample (b) with particle size distribution in inset; c SAED pattern of sample (a); d SAED pattern of sample (b)
The 13C resonance signals have been assigned according to chemical shift theory. The considerable shielding (~ 2–3 ppm) takes place in the position of C–O and C=N in spectra of the above complexes as compared to ligands, indicating coordination through the oxigen and the nitrogen to metal. These data corroborate the observations obtained from the IR spectral studies. In the 13C NMR spectra, all the other signals are appeared at the expected regions.
3.1.3 Thermal Studies
The thermogravimetric analysis (TG) of representative precursor [PdCl2·C15H12Cl2N2O](1) was carried out nitrogen atmosphere (Fig. 4, Supplementary Materials). The onset decomposition of precursor was started at 170 °C and it decomposed in different steps due to multiple weight loss steps and organic pyrolys is events. The decomposition appears to be completed at about 800 °C. Here PdO was obtained as a final product [28.49% (29.78%)].

HRTEM images of: a sample (a) showing core and shell parts, b sample (b)
3.2 Sol–Gel Transformations
Precursors Sol–gel transformations of [PdCl2·C15H12Cl2N2O](1) and PdCl2 (2) to pure Pd@PdO core shell nanoparticles (a) and PdO nanoparticles (b), respectively have been carried by the method shown in Fig. 1. The method presented here is more simple and versatile than other reported methods. Ruffino et al. synthesized core–shell nanoparticles via laser irradiation of Au films on Si wafer [31]. Choma et al. synthesize silica and gold nanoparticles separately and then did modification by adding surface organic groups on silica particles to adhere gold nanoparticles to form a core–shell structure which is very tedious route [32]. Fratoddi et al. have shown multi-step rigorous process to synthesize metal/polymer core–shell nanoparticles [33]. Unlike these methods, we have presented a simple process of precursor synthesis and sol–gel transformation of precursor into core–shell nanoparticles. The method gives freedom to synthesize metal oxide nanoparticles and metal/metal oxide core–shell nanoparticles by just modifying the precursors. The precursor serves as the source of the elements of desired core–shell nanoparticles. The sol–gel method is based on the hydroxylation and condensation of molecular precursors in a solution, originating a sol of nanometric particles. Condensation and inorganic polymerization of the sol lead to the formation of a gel made of a three-dimensional metal oxide network (Reddy et al. [34]) [34]. The crystalline state will be obtained then upon heat treatment of the gel. The control over the characteristics and structure of the gel can be easily obtained by fixing the hydroxylation and condensation conditions and the kinetic parameters of the growing process (such as pH, temperature, nature and concentration of the salt precursors, solvent nature, precursor structure) (Itoh and Sugimoto [35]) [35]. The structure of the final product can be designed (particle, nanorods, core–shell etc.) by changing these parameters [36]. In our case, we have just used modification in precursor structure to obtain core–shell structure in synthesized nanoparticles. During the sol–gel synthesis, all elements present in the ionic form where Once Pd(0) formed, dissociative adsorption of oxygen takes place on the surface of the particles. This is followed by the diffusion of oxygen atoms into the Pd lattice by forcing apart the Pd atoms to give PdO at > 100 °C. Complete oxidation of Pd(0) to PdO was not observed in our case and this could be due to the diffusion of oxygen only on to the surface layers. The PdO layer was found to be quite stable, heating the core–shell structure at higher temperatures did not result in the oxidation of the core which suggested that PdO formed a good protective shell over the Pd core [16, 37].
The X-ray diffraction patterns of synthesized samples (a) and (b) are shown in Fig. 2. A comparative study of the XRD patterns of samples (a) and (b), indicate the predominant formation of PdO (tetragonal phase) for (b). In case of sample (a), cubic phase was generated in the presence of tetragonal phase which shows the synthesis of Pd@PdO. In the XRD pattern of sample (b), reflections peaks at 2θ = 33.75, 41.80, 54.61, 60.06 and 60.65, corresponding to the pure tetragonal phase of PdO, and could be indexed to the (100), (101), (110), (112), (103) and (200) planes (JCPDS#85-0713) [16]. The XRD pattern of the sample (a), indicated both the formation of cubic (fcc) Pd and tetragonal PdO phase; reflections from the (111), (200) and (220) planes of Pd(0) and (101), (110), (112), (103) and (200) planes of PdO were observed (JCPDS # 46-1043 and 85-0713) [16]. The lattice parameters calculated were a = 3.02 Å b = 3.02 Å c = 5.31 Å for sample (a and b) (tetragonal) and a = b = c = 3.89 Å for Pd of Pd@PdO of sample (a) (cubic). Complete oxidation of Pd to PdO did not take place even at higher temperatures (600 °C) suggesting a core–shell structure consisting of a shell of PdO protecting a Pd(0) core (blue trace, Fig. 2). The average crystallite size in samples {33 ± 3 nm for (a), and 25 ± 3 nm for (b)} was calculated, by using the Debye–Scherrer equation [38].
Figure 3 shows the TEM images and corresponding SAED patterns of both the samples. The TEM image of sample (a) shows the presence of nanoparticles. Corresponding SAED pattern shows presence of planes for both Pd and PdO, suggesting formation of core–shell structure. The particle size distribution (inset of Fig. 3a) shows variation of particles size from 40 nm to 100 nm for sample (a) with the average particle size of 70 nm and standard deviation of 18. The TEM image of the sample (b) indicates formation of polygonal shaped particles. Corresponding SAED pattern shows the presence of PdO planes. The particle size distribution (inset of Fig. 3b) shows variation of particle size from 20 to 90 nm with average particle size of 55 nm and standard deviation of 10. SAED pattern of (a) and (b) shows that both the samples are of polycrystalline nature and the results are well matched with XRD results.
The HRTEM image of sample (a) clearly shows the presence of core–shell nanoparticles (Fig. 4a) which is not the case in sample (b) as shown in Fig. 4b. The size of core part and shell is 36 ± 2 nm and 3.5 ± 0.5 nm, respectively. So the ratio between core and shell is of about 9:1. The ratio and size of core and shell part significantly affect the light scattering thus optical properties of the core–shell nanoparticles [39, 40]. The d-spacing measured by HRTEM and the planes obtained by SAED patterns were corroborated with the XRD results of the samples. The Fig. 5 shows HRTEM image with point SAED patterns. It can be seen in Fig. 5 that both PdO and Pd phases are present in the single particle. Thus the combined powder XRD and electron microscopy results indicated the formation of a core–shell structure of Pd@PdO for sample (a) and tetragonal for (b).

HRTEM image of sample (a) showing presence of PdO (in black) and Pd (in red). (Color figure online)
The surface morphology of the samples (a) and (b), observed by scanning electron microscopy are shown in Fig. 6. The images indicate agglomerated granular morphologies for both samples, (a) and (b) with nano sized particles. The homogeneous and good quality of the particles with almost consistent diameter can be seen in the SEM image, shows the trustworthiness of the method. In order to investigate the composition materials, EDX analysis (Fig. 6) of these samples were also carried out indicating the formation of PdO and Pd@PdO.

SEM images of: a sample (a), b sample (b); EDX patterns of: c sample (a), d sample (b)
A typical full Raman spectra from a PdO and Pd@PdO samples obtained by sol gel synthesis were recorded at room temperature is displayed in Fig. 7. The Pd as metallic nanoparticles has no individuality active Raman modes while PdO is highly Raman-active material. More exactly, and on the basis of the reported literature, it is confirms that PdO is strongly detectable by Raman scattering spectroscopy even without the need for an external enhancement approach [8]. This is attributed to its resonant Raman scattering property.

Raman spectra of sample (a) (in blue) and (b) (in red). (Color figure online)
As shown in Fig. 7, three relatively intense Raman modes are observed at the following Raman shifts: 647 cm−1, 424 cm−1and 275 cm−1 which assigned to the B1g, Eg and X8 modes respectively [41, 42]. Commonly, PdO has two Raman active modes B1g and Eg which are related to theunit cell of the tetragonal phase of PdO crystal [41, 42]. The Raman modes were observed in both the samples as PdO is also present in sample (a) which also confirms the presence of PdO at the surface of sample (a). A little shift in the peak positions can be seen in Fig. 7 as compared to the earlier reports which can be attributed to nano size and presence of stresses in the samples.
In the FT-IR spectra of PdO samples, the absorption bands observed around at 606–608 cm−1 and at 661–666 cm−1, appeared due to the Pd–O vibrations. These peaks was further supported the formation of PdO in both the samples [30]. The ν(–OH) absorption in the region of 3430–3439 and 1631–1652 cm−1 indicate the stretching and bending vibrations, due to the absorption of moisture [43].
The UV–Visible absorption spectra of both the samples are shown in Fig. 8. Optical bandgap for both the samples (a) and (b) were extrapolated using the Tauc relation [44]. Plots of hν versus (αhν)2 (Fig. 9) of the samples (a) and (b) exhibit energy band gaps 1.43 eV, and 1.89 eV, respectively. The bandgap of the Pd@PdO nanoparticles is very near to the optimal bandgap (~ 1.5 eV) of absorber layer in the solar cell [45] which suggests its possible use in solar applications. It has been reported previously that the bandgap of the material is inversely proportional to the particle and crystallite size of the material [46, 47]. So here, increase in the energy band gap can be attributed to the re-arrangement of the energy bands due to reduction in particle and crystallite size.

UV–Visible spectra of sample (a) and (b)

Tauc plots of samples (a) and (b)
4 Conclusions
Sol–gel transformations can be used to synthesize Pd@PdO core–shell nanoparticles. Sol–gel transformation of PdCl2 andPdCl2·C15H12Cl2N2O into tetragonal PdO and Pd@PdO core–shell make these complexes suitable for the synthesis of nano structure, respectively. Bandgap of Pd@PdO nanoparticles near to the optimal bandgap of absorber layer in a solar cell suggests a new future application of the palladium nanoparticles.
References
J. Tsuji, Palladium Reagents and Catalysts New Perspectives for the 21st Century (Wiley, Chichester, 2004)
R. Narayanan, M.A. El-Sayed, Carbon-supported spherical palladium nanoparticles as potential recyclable catalysts for the Suzuki reaction. J. Catal. 234, 348–355 (2005). https://doi.org/10.1016/j.jcat.2005.06.024
O.M. Wilson, M.R. Knecht, J.C. Garcia-Martinez, R.M. Crooks, Effect of Pd nanoparticle size on the catalytic hydrogenation of allyl alcohol. J. Am. Chem. Soc. 128, 4510–4511 (2006)
J. Huang, Y. Liu, H. Hou, T. You, Simultaneous electrochemical determination of dopamine, uric acid and ascorbic acid using palladium nanoparticle-loaded carbon nanofibers modified electrode. Biosens. Bioelectron. 24, 632–637 (2008). https://doi.org/10.1016/j.bios.2008.06.011
D. Sil, J. Hines, U. Udeoyo, E. Borguet, Palladium nanoparticle-based surface acoustic wave hydrogen sensor. ACS Appl. Mater. Interfaces 7(10), 5709–5714 (2015). https://doi.org/10.1021/am507531s
N.R. Jana, Z.L. Wang, T. Pal, Redox catalytic properties of palladium nanoparticles: surfactant and electron donor—acceptor effects. Langmuir 16, 2457–2463 (2000). https://doi.org/10.1021/la990507r
L. Liu, S.-H. Yoo, S.A. Lee, S. Park, Wet-chemical synthesis of palladium nanosprings. Nano Lett. 11, 3979–3982 (2011). https://doi.org/10.1021/nl202332x
J.T. Remillard, W.H. Weber, J.R. McBride, R.E. Soltis, Optical studies of PdO thin films. J. Appl. Phys. 71, 4515–4522 (1992). https://doi.org/10.1063/1.350797
E. Kanezaki, S. Tanaka, K.-I. Murai, T. Moriga, J. Motonaka, M. Katoh, I. Nakabayashi, Direct detection of a phase change in PdO/CeO2 supported on χ-Al2O3 by Means of in situ high-temperature measurements of XRD and FTIR. Anal. Sci. 20, 1069–1073 (2004). https://doi.org/10.2116/analsci.20.1069
K.R. Thampi, J. Kiwi, M. Gratzel, Oxidative dimerisation of methane on supported palladium oxide catalysts. Catal. Lett. 4, 49–56 (1990). https://doi.org/10.1007/BF00764870
Y. Ozawa, Y. Tochihara, M. Nagai, S. Omi, PdO/Al2O3 in catalytic combustion of methane: stabilization and deactivation. Chem. Eng. Sci. 58, 671–677 (2003). https://doi.org/10.1016/S0009-2509(02)00594-8
T.L. Stuchinskaya, I.V. Kozhevnikov, Liquid-phase oxidation of alcohols with oxygen catalysed by modified palladium(II) oxide. Catal. Commun. 4, 417–422 (2003). https://doi.org/10.1016/S1566-7367(03)00096-7
X. Xu, W. Yang, J. Liu, L. Lin, Synthesis of a high-permeance NaA zeolite membrane by microwave heating. Adv. Mater. 12, 195–204 (2000). https://doi.org/10.1002/(SICI)1521-4095(200002)12:3%3C195::AID-ADMA195%3E3.0.CO;2-E
E. Ismail, M. Khenfouch, M. Dhlamini, S. Dube, M. Maaza, Green palladium and palladium oxide nanoparticles synthesized via Aspalathus linearis natural extract. J. Alloy. Compd. 695, 3632–3638 (2017). https://doi.org/10.1016/j.jallcom.2016.11.390
B.L. Justus, R.J. Tonucci, A.D. Berry, Nonlinear optical properties of quantum confined GaAs nanocrystals in Vycor glass. Appl. Phys. Lett. 61, 3151–3153 (1992)
D. Jose, B.R. Jagirdar, Synthesis and characterization of Pd(0), PdS, and Pd@PdO core–shell nanoparticles by solventless thermolysis of a Pd–thiolate cluster. J. Solid State Chem. 183, 2059–2067 (2010). https://doi.org/10.1016/j.jssc.2010.07.013
C.J. Huang, F.M. Pan, T.C. Tzeng, L. Chan, J.T. Sheub, Growth and field emission of reactive sputtered Pd–PdO core–shell nanoflakes on platinum. J. Electrochem. Soc. 156(2), J28–J31 (2009). https://doi.org/10.1149/1.3035822 https
B. Jiang, S. Song, J. Wang, Y. Xie, W. Chu, H. Li, X.H. Chungui Tian, H. Fu, Nitrogen-doped graphene supported Pd@PdO core–shell clusters for C–C coupling reactions. Nano Res. 7(9), 1280–1290 (2014). https://doi.org/10.1007/s12274-014-0492-1
S. Sakka, Handbook of Sol-Gel Science and Technology: Characterization and Properties of Sol-Gel Materials and Products (Springer, Switzerland, 2005)
V.G. Kessler, G.I. Spijksma, G.A. Seisenbaeva, S. Hakansson, D.H.A. Blank, H.J.M. Bouwmeester, New insight in the role of modifying ligands in the sol–gel processing of metal alkoxide precursors: a possibility to approach new classes of materials. J. Sol-Gel Sci. Technol. 40, 163–179 (2006). https://doi.org/10.1007/s10971-006-9209-6
D. Avnir, T. Coradin, O. Lev, J. Livage, Recent bio-applications of sol–gel materials. J. Mater. Chem. 16, 1013–1030 (2006). https://doi.org/10.1039/B512706H
A.E. Danks, S.R. Hall, Z. Schnepp, The evolution of ‘sol–gel’ chemistry as a technique for materials synthesis. Mater. Horiz. 3, 91–112 (2016). https://doi.org/10.1039/C5MH00260E
F.L. Li, H.J. Zhang, Synthesis of hollow sphere and 1D structural materials by sol-gel process. Materials 10, 995–1011 (2017). https://doi.org/10.3390/ma10090995
L.L. Hench, J.K. West, The sol-gel process. Chem. Rev. 90, 33–72 (1990). https://doi.org/10.1021/cr00099a003
C.J. Brinker, G.W. Scherer, Sol–Gel Science: The Physics and Chemistry of Sol–Gel Processing (Academic Press, California, 1990)
M.K. Atal, R. Sharma, A. Saini, V. Dhayal, M. Nagar, Synthesis and characterization of organotin(IV) semicarbazones: potential precursors for nanosized tin oxide. J. Sol-Gel Sci. Technol. 79(1), 114–121 (2016). https://doi.org/10.1007/s10971-016-4013-4
A.I. Vogel, A Text Book of Quantitative Inorganic Analysis, 5th edn. (Longman, London, 1989)
R.A. Muna, A. Al-Mandhary, P.J. Steel, 8-(2-pyridylmethoxy)quinoline: a new N,O,N?-tridentate ligand and X-ray crystal structures of its mononuclear palladium(II), copper(II) and nickel(II) complexes. Inorg. Chim. Acta. 351, 7–11 (2003). https://doi.org/10.1016/S0020-1693(03)00188-9
A.M. Tajuddin, H. Bahron, K. Kassim, W. Nazihah, W. Ibrahim, B.M. Yamin, Synthesis and characterization of paladium (II) Schiff base complexes and their catalytic activities for Suzuki coupling reaction. Malays. J. Anal. Sci. 16(1), 79–87 (2012)
M.J. Tura, P. Regull, L. Victorit, D.D. Castellar, XPS and IR (ATR) analysis of Pd oxide films obtained by electrochemical methods. Surf. Interface Anal. 11, 447–449 (1988). https://doi.org/10.1002/sia.740110807
F. Ruffino, A. Pugliara, E. Carria, L. Romano, C. Bongiorno, C. Spinella, M.G. Grimaldi, Novel approach to the fabrication of Au/silica core-shell nanostructures based on nanosecond laser irradiation on thin Au films on Si. Nanotechnology 23, 045601 (2012)
J. Choma, A. Dziura, D. Jamioła, P. Nyga, M. Jaroniec, Preparation and properties of silica–gold core–shell particles. Colloids Surf. A 373, 167–171 (2011)
I. Fratoddi, I. Venditti, C. Battocchio, G. Polzonetti, C. Cametti, M.V. Russo, Core shell hybrids based on nobel metal nanoparticles and conjugated polymers: synthesis and characterization. Nanoscale Res. Lett. 6, 98 (2011)
L.H. Reddy, J.L. Arias, J. Nicolas, P. Couvreur, Magnetic nanoparticles: design and characterization, toxicity and biocompatibility, pharmaceutical and biomedical applications. Chem. Rev. 112 (11), 5818–5878 (2012). https://doi.org/10.1021/cr300068p
H. Itoh, T. Sugimoto, Systematic control of size, shape, structure, and magnetic properties of uniform magnetite and maghemite particles. J. Colloid Interface Sci. 265(2), 283–295 (2003). https://doi.org/10.1016/S0021-9797(03)00511-3
R. Gopal, A. Goyal, A. Saini, M. Nagar, N. Sharma, D.K. Gupta, V. Dhayal, Sol-gel synthesis of Ga2O3 nanorods and effect of precursor chemistry on their structural and morphological properties. Ceram. Int. 44, 19099–19105 (2018)
E. Lundgren, J. Gustafson, A. Mikkelsen, J.N. Andersen, A. Stierle, H. Dosch, M. Todorova, J. Rogal, K. Reuter, M. Scheffler, Kinetic hindrance during the initial oxidation of Pd(100) at ambient pressures. Phys. Rev. Lett. 92, 046101/1–046101/4 (2004). https://doi.org/10.1103/PhysRevLett.92.046101
B.E. Warren, X-Ray Diffraction (chap. 13) (Dover Publication, New York, 1990)
F. Ruffino, A. Pugliara, E. Carria, C. Bongiorno, M.G. Grimaldi, Light scattering calculations from Au and Au/SiO2 core/shell nanoparticles. Phys. E 47, 25–33 (2013)
A.I. Barreda, Y. Gutierrez, J.M. Sanz, F. Gonzalez, F. Moreno, Light guiding and switching using eccentric core-shell geometries. Sci. Rep. 7, 11189 (2017). https://doi.org/10.1038/s41598-017-11401-y
R. Bardhan, A. Schwartzberg, C.L. Pint, Size-dependent phononic properties of PdO nanocrystals probed by nanoscale optical thermometry. J. Phys. Chem. C 117, 21558–21568 (2013). https://doi.org/10.1021/jp406916h
G.W. Graham, D. Uy, W.H. Weber, H. Sun, X.Q. Pan, Observation of strained PdO in an aged Pd/ceria-zirconia catalyst. Catal. Lett. 79, 99–105 (2002). https://doi.org/10.1023/A:1015344301416
X. Wei, D. Chen, Synthesis and characterization of nanosized zinc aluminate spinel by sol–gel technique. Mater. Lett. 60, 823–827 (2006). https://doi.org/10.1016/j.matlet.2005.10.024
J Tauc, Amorphous & Liquid Semiconductors (Plenum, New York, 1974)
W. Shockley, H.J. Queisser, Detailed balance limit of efficiency of p-n junction solar cells. J. Appl. Phys. 32, 510–519 (1961). https://doi.org/10.1063/1.1736034
A. Goyal, V. Sharma, A. Sharma, R. Agarwal, K.B. Sharma, S.L. Kothari, Synthesis, structural and optical study of CdS nanoparticles doped with different concentration of Cu. J. Nano Electron. Phys. 3, 254 (2011)
W.L. Wilson, P.J. Szajowski, L.E. Brus, Quantum confinement in size-selected, surface-oxidized silicon nanocrystals. Science 262, 1242–1244 (1993). https://doi.org/10.1126/science.262.5137.1242
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kumari, T., Gopal, R., Goyal, A. et al. Sol–Gel Synthesis of Pd@PdO Core–Shell Nanoparticles and Effect of Precursor Chemistry on Their Structural and Optical Properties. J Inorg Organomet Polym 29, 316–325 (2019). https://doi.org/10.1007/s10904-018-1001-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10904-018-1001-x