
Abstract
The present research work is a systematic study to know the influence of Mn doping on structural, electronic, and optical properties of ZnTe nanoclusters (NCs). The calculations are performed on 4-atom, 6-atom, 8-atom, and 14-atom molecules of pure and Mn-doped ZnTe using Generalized Gradient Approximation (GGA) in the framework of density functional theory implemented in Amsterdam Density Functional (ADF). It has been found that 6-atom (Zn3Te3) has hexagon while 8-atom (Zn4Te4) Zinc-blende structure with TD symmetry and the smallest units of ZnTe NCs. The Raman and IR spectra are also studied, which are comparable to the reported theoretical results. Different modes such as symmetric, Asymmetric, scissoring, rocking, wagging, and twisting are identified. Due to quantum size affecting the wavelengths of the calculated absorption peaks gradually shift to red in order of 4- atom, 6-atom, 8-atom, and 14-atom.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The II-VI compound semiconductors at nanoscale are of great interest because of their wide direct bandgap, stability of fundamental structural, electrical, and optical properties. They have many potential applications in thin-film solar cells, photo-detectors, photocatalytic and optoelectronic devices [1, 2]. Substitution of transition metals (Mn, Cr, Fe, Ni, Co etc) into non-magnetic semiconductor (II-VI, II-V, III-V and IV-VI) gives rise to new class of semiconducting materials known as dilute magnetic semiconductors which are the most auspicious materials for applications in photo-catalytic and spintronic devices [3, 4]. The II-VI compound DMSs such as ZnTe, ZnSe, CdSe and CdTe, magnetic ions are easily absorbed with +2 oxidation states [5]. Mn-doped DMSs provide a few imperative applications and show anti-ferromagnetic property while Cr-doped DMSs show ferromagnetism [6, 7]. Zinc Telluride (ZnTe) is II–VI semiconductor with band gap 2.21 to 2.26 eV at room temperature and high optical absorption coefficient, show zinc-blende structure, has many potential applications in different types of solid state devices such as solar cells, photo-detectors and light emitting diodes [8]. Literature survey shows that ZnTe has been studied extensively and found its uses in hetero-junction solar cells as a back contact for CdTe, visible light emitting semiconductor laser, substrate for the growth of CdTe and hetero-structures based on ZnTe and HgTe are used for infrared optics [9,10,11,12]. It is used for optical power limiting applications on doping with vanadium [13]. This material can minimize the toxic nature by exchanging chemical bath deposited CdS layers when it is used as a window layer in thin film solar cells [14]. Remarkable change in optical, electrical, and mechanical properties of CdTe/ZnTe thin films is observed by doping. In order to increase the conductivity of ZnTe thin films it is doped with Ag impurity. Similarly, copper, nitrogen and sodium doped ZnTe has important application in CdTe solar cells as stable electrical back contacts [15]. Copper doped ZnTe becomes very important in the research due to its wide range of applications in microelectronics and optoelectronics [16, 17]. ZnTe NCs has been studied recently by Xu et al. under DFT but they did not identify the modes of vibrations [18]. In this work we have identified all modes of vibrations like symmetric, asymmetric, scissoring, rocking, wagging and twisting. Moreover, the theoretical study of Mn doped ZnTe is not available in literature at nanoscale, this article has tried to fill this deficit, results of pure and doped ZnTe are discussed.
Computational Details
Amsterdam Density Functional (ADF) [19] and Vienna Ab-Initio Simulation (VASP) [20] packages are used to perform calculations depending upon Kohn-Sham density functional approach. The Generalized Gradient Approximation (GGA) exchange and correlation function with the parameterization of Perdew Burke Ernzerhof (PBE) functional is evaluated within DFT. We had generated these structures with ab-initio random structure searching (AIRSS) [21]. Initially these structures were optimized with, and further calculations, such as geometric optimization, Raman and IR spectra were performed with ADF. The Quadruple zeta double polarization (QZ2P) basis sets are used for all calculations. The GGA-PBE and QZ2P basis sets predict the best results in analyzing the molecular geometries, enthalpies and vibrational properties [22]. The 4-atom, 6-atom, 8-atom and 14-atom of pure and Mn doped ZnTe are simulated in ADF-GUI. The valence electronic configurations are Zn (3d104s2) and Te (5s25p4) and the Zn-4d electrons or core electrons in the valence configuration are considered as inert [23]. The accuracy parameter is set to 4. The geometry optimization and other calculations of these NCs have been performed with energy convergence criteria of 10-5eV, gradient convergence criteria of 10-5 eV/Å and radial convergence criteria of 10-2 Å. Atomic orbital having similar energies and the appropriate symmetry can be combined to form molecular orbital [24]. Hydrogen has been used as ligand because it saturates the NCs to overcome the open shell calculations and dangling bond problems.
Result and Discussion
This section provides the information about the structural, optical, and electronic properties of 4, 6, 8 and 14-atom pure and Mn-doped ZnTe NCs. Varying number of atoms affect the band gap of NCs, it decreases with increasing number of atoms. The IR and Raman spectrum of all the NCs are calculated and the peaks observed in each spectra are compared with the experimental data (available in literature [25,26,27]).
Structural Properties
The binding energy with increasing size of NCs, EB, is calculated as: [28]
And with Mn doping
A higher positive value of EB corresponds to a higher stability of the cluster.
Figure. 1shows the binding energy of ZnTe (black) and Mn-ZnTe (Red). The stability of each NCs increases with doping of Mn-atom. The 10-atom structure is less stable while 4-atom,

The binding energy vs. size of the ZnnTen and Znn-MnTen, (n = 1–8) NCs (Color figure online)
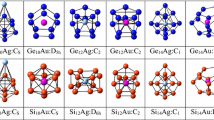
6-atom, 8-atom 12 and 14-atom clusters are stable. The 12-atom cluster is ignored because we need to dope these NCs with Mn-atom. In case of 4-atom, 6-atom and 8-atom, there is only one choice to dope Mn due to high symmetry. To retain high symmetry for active site, the14-atom NCs is chosen instead of 12-atom. In 12-atom NC, there are two hexagonal rings, so doping at edge could break symmetry as in 6-atom Mn doped (Fig. 2A(d)). Overall, the idea behind considering 4-atom and 8-atom is because these are squared or cubic NCs structures, while 6-atom and 14-atom NCs are hexagonal structures. These high symmetry structures based on their shape and daggling bonds are chosen for Raman and IR spectra. This choice gives a comparative information between 4 and 8-atom and 6 and 14-atom from these spectra. Figure 2 shows the [A] Molecular Structures and [B] Contour plots of 4-atom, 6-atom, 8-atom, and 14-atom of pure and Mn-doped ZnTe NCs. The bond lengths, energy gap, and bonding energies of pure ZnTe and Mn-ZnTe of 4, 6, 8 and 14-atom are given in Table 1. The energy gaps of pure ZnTe of 6, 8 and 14-atom is closely related to experimental value of 2.26 eV [29].

[A] Molecular Structures and [B]Contour Plots of Pure and Mn-Doped ZnTe (Color figure online)
The Zn-Te bond lengths for 4, 6, 8 and 14-atom show decreasing trend while Mn-Te show increasing trend and the Mn-H bond lengths are remained constant. There is no significant difference in the crystal structure of doped and un-doped clusters as well as bond lengths, but energy gaps are decreased in doped NCs as compared to un-doped. The 4-atom structure is planar, and 6-atom is hexagonal ring. The structure of 8-atom ZnTe is challenging structure having similarity with zinc-blende structure. The bond length between the atoms of Zn and Te is 2.786 Å. The 6-atom NCs can be modified to large clusters with the increase in number of hexagons as we have simulated 14-atom NCs which is also hexagonal. Moreover, contour plots in Fig. 2[B] show that bonding between Zn-Te is ionic. Furthermore, the values of entropy, binding energies and zero-point energies given in Table. 1 show that all NCs are thermodynamically stable, and this stability increases with the increase in the size of NCs.
Electronic Properties
Molecular orbitals i.e, HOMO and LUMO along with their properties are very useful for physicist and chemist and are used by the frontier electron density. The HOMO represents the capability to accommodate an electron (Ionization potential) whereas LUMO as an electron acceptor to characterize the capability to accept an electron (Electron affinity) (Fig. 3).

Energy Level Diagrams of Pure and Mn doped ZnTe
The central aspect of the electronic properties is the energy gap between occupied and unoccupied electron states, the HOMO-LUMO (H-L) gap. In small clusters quantum effect becomes important and as a result large energy gap has been appeared. The increase in cluster’s size, quantum effect becomes less important and thus larger size clusters would have insignificant energy gap. The HOMO and LUMO are also very important in approximating stability, determining transition, and investigating type of material. The clusters with large H-L gaps are more stable as compared to those with lower values of the band gap. As the cluster size increases, H-L gap increases, this leads to a less chemical reactivity. To estimate the electronic stability of small clusters H-L gap is an important parameter. The large energy gap has a direct relationship with electronic stability and inverse with reactivity and vice versa. Moreover, the values of bonding energy (eV) in Table. 1 show that NCs become thermodynamically stable as the size of these clusters increases. The 8-atom ZnTe is a fascinating high symmetry (Td) zinc-blende structure. The calculated HOMO and LUMO is delocalized over the whole cluster, meaning that saturation of dangling bonds abstains from the appearance of surface localized trap states. The HOMO is mostly affirmed over the anion p-valence orbital, while LUMO is mostly composed of Zn-4s orbital. The H-L gap values of pure ZnTe and Mn doped ZnTe for 4-atom, 6-atom, 8-atom and 14-atom are given in Table 1. We have observed that the band gap reduces significantly on doping. Furthermore, we can adjust H-L gap by changing the number of atoms. This rule is induced by quantum size effect.
To further study molecular orbital characters, we generated HOMO and LUMO of these structures. Fig. 4a, c, e, and g shows the HOMO-LUMO plots of pure ZnTe while Fig. 4b, d, f, and h shows HOMO and LUMO of Mn doped ZnTe. Obviously, there is d orbital in HOMO and there is no d in LUMO which is composed of p orbital. Moreover, all absorption peaks are transitions from HOMO to LUMO. It testifies that all transitions in these structures are from d to p-orbital. Namely, d-orbital of Te-atom does not participate in bonding. Besides, seeing from HOMO–LUMO plots in Fig. 4, we can find that 4-atom ZnTe structure has strong delocalization which provides it better stability that is important for structure formation experimentally.

HOMO–LUMO plots of Pure (left) and Mn-doped ZnTe (right) (Color figure online)
Raman and Infrared (IR) Spectroscopy
To find the vibrational and structural information of ZnTe NCs, we have calculated Raman and IR spectra of 4-atom, 6-atom, 8-atom and 14-atom of pure and Mn doped NCs and are given in Fig. 5. The modes of vibrations of these NCs are given in Table 2.

Raman Spectra of 4, 6, 8 and 14-atom Pure and Mn-doped ZnTe (Color figure online)
The above-mentioned peaks represent different modes of vibrations i.e, symmetric and asymmetric stretching, bending, or scissoring, twisting, rocking, and waging at frequencies as listed in Table. 2.
In case of Mn doped ZnTe NCs, we saturate them with hydrogen to make the close shell configuration and to introduce the solvent environment. The observed Raman peaks for Mn- doped ZnTe with 4-atom, 6-atom, 8-atom and finally 14-atom Mn : ZnTe NC are also given in Table 2. In addition to these peaks, some peaks for hydrogen are observed at high frequencies i.e, 1850, 1882, and 1917 cm-1 [18]. These results are comparable with experimental results (126, 175, 204, and 406 cm−1; modes of vibrations for pure ZnTe). The Raman peaks at 177 and 206 cm-1 are attributed to the transverse optic and longitudinal optic phonon modes of ZnTe, respectively [26]. Doping of ZnTe with transition metals and saturation of hydrogen provides IR spectra with greater number of peaks and frequencies. Different IR active modes of vibrations are shown in Fig. 6.

IR Spectra of 6, 8 and 14-Atom Pure and Mn-doped ZnTe
Finally, different modes of vibrations are investigated at above mentioned peaks in Fig. 7.

Modes of vibrations for a pure ZnTe and b Mn-doped ZnTe (Color figure online)
It is absorbed that if the frequency of modes of vibration increases with increasing cluster size, it is blueshift and if the frequency of modes of vibration decreases with increase in cluster size it is the redshift. In pure ZnTe and Mn-ZnTe, the symmetric mode shows blueshift while asymmetric mode of vibration shows redshift. The twisting, symmetric and asymmetric modes show redshift while remaining show blueshift in Mn:ZnTe while in pure ZnTe, the symmetric modes show blueshift and asymmetric modes show redshift. It can be seen that symmetric, asymmetric, Scissoring and twisting are present for all NCs. But rocking mode is absent for 8-atom and wagging mode is absent for 4 and 8-atom. These modes strongly depend on the structure of NCs. This investigation clearly shows that 6-atom and 14-atom have same (hexagonal) structure as all modes of vibrations are present. Moreover, 4-atom (Planer) is different from 8-atom (Zinc-blende) due to presence of rocking mode in 4-atom NC.
Conclusion
The purpose of this research work is to investigate the properties of 4, 6, 8, and 14-atom ZnTe NCs. The energy gap of ZnTe NCs can be varied by the substitution of dopant atom and increasing the number of atoms in a cluster. The electronic and optical properties depend upon the size of the NCs due to quantum confinement effect in nano-materials. The electronic and thermodynamic stabilities increase with the increase in size of NCs. Raman and IR analysis of pure and Mn-doped ZnTe clearly explain the modes of vibrations at different frequencies and as well different structures of these NCs.
References
S. Jeetendra, H. Nagabhushana, K. Mrudula, C. S. Naveen, P. Raghu, and H. M. Mahesh (2014). Concentration dependent optical and structural properties of Mo doped ZnTe thin films prepared by e-beam evaporation method. Int. J. Electrochem. Sci. 9, 2944–2954.
D. G. Diso, F. Fauzi, O. K. Echendu, A. R. Weerasinghe, and I. M. Dharmadasa (2011). Electrodeposition and characterisation of ZnTe layers for application in CdTe based multi-layer graded bandgap solar cells. J Conf Ser Phys. https://doi.org/10.1088/1742-6596/286/1/012040.
H. Ohno (1998). Making nonmagnetic semiconductors ferromagnetic. Science (80- ) 281, 951–956.
A. Haury, A. Wasiela, A. Arnoult, J. Cibert, S. Tatarenko, T. Dietl, and Y. Merle d’aubigné (1997). Observation of a ferromagnetic transition induced by two–dimensional hole gas in modulation–doped CdMnTe quantum wells. Phys. Rev. Lett. 79, 511–514.
A. You, M. A. Y. Be and I. In, New III-V diluted magnetic semiconductors ( invited ) New Ill-V diluted.
M. Rigana Begam, N. M. Rao, G. M. Joshi, S. Kaleemulla, M. Shobana, N. Sai Krishna, and M. Kuppan (2013). Structural, optical, and magnetic properties of co doped cdte alloy powders prepared by solid-state reaction method. Adv. Condens. Matter Phys. 2013, 2–7.
J. K. Furdyna (1982). Diluted magnetic semiconductors: An interface of semiconductor physics and magnetism (invited). J. Appl. Phys. 53, 7637–7643.
C. X. Shan, X. W. Fan, J. Y. Zhang, Z. Z. Zhang, X. H. Wang, J. G. Ma, Y. M. Lu, Y. C. Liu, D. Z. Shen, X. G. Kong, and G. Z. Zhong (2002). Structural and luminescent properties of ZnTe film grown on silicon by metalorganic chemical vapor deposition. J. Vac. Sci. Technol. A Vacuum, Surfaces, Film 20, 1886.
M. Imamura, A. Okada, and T. Yamaguchi (2006). Magneto-optical properties of wider gap II-VI ZnMnTe and ZnMnCoTe films. J. Appl. Phys. 99, 1–4.
S. A. Dvoretsky, D. G. Ikusov, Z. D. Kvon, N. N. Mikhailov, V. G. Remesnik, R. N. Smirnov, Y. G. Sidorov, and V. A. Shvets (2008). HgCdTe quantum wells grown by molecular beam epitaxy. Semicond. Physics, Quantum Electron. Optoelectron. 10, 47–53.
V. Kumar, G. S. Sandhu, T. P. Sharma, and M. Hussain (2007). Growth and characterization of Cd1−XZnX Te-sintered Films. Res. Lett. Mater. Sci. 2007, 1–5.
L. P. Deshmukh, K. M. Garadkar, and D. S. Sutrave (1998). Studies on solution grown HgXCd1 - XS thin films. Mater. Chem. Phys. 55, 30–35.
S. Tobeñas, E. M. Larramendi, E. Purón, O. De Melo, F. Cruz-Gandarilla, M. Hesiquio-Garduño, and M. Tamura (2002). Growth of Cd(1–x)ZnxTe epitaxial layers by isothermal closed space sublimation. J. Cryst. Growth 234, 311–317.
J. L. Ecuyer, N. Audet, D. Shink, R. Triboulet, K. W. Benz, M. Fiederle, D. Lincot, V. N. Tomashik and Z. F. Tomashik, Crystal Growth and Surfaces, 2010.
T. M. Razykov (1988). Physical properties of II-VI binary and multi-component compound films and heterostructures fabricated by chemical vapour deposition. Thin Solid Films 164, 301–308.
A. Kindvall, A. Munshi, T. Shimpi, A. Danielson and W. S. Sampath, Copper-Doped Zinc Telluride Thin-Films as a Back Contact for Cadmium Telluride Photovoltaics, 2018 IEEE 7th World Conf. Photovolt. Energy Conversion, WCPEC 2018 - A Jt. Conf. 45th IEEE PVSC, 28th PVSEC 34th EU PVSEC, 2018, 2994–2997.
S. H. Cho, J. H. Suh, J. H. Won, K. H. Kim, J. K. Hong, and S. U. Kim (2008). Surface leakage current control with heterojunction-type passivation in semi-insulating CdZnTe material. Methods Phys. Res. Sect. A Accel. Spectrometers, Detect. Assoc. Equip. 591, 203–205.
S. Xu, C. Wang, and Y. Cui (2009). Influence of ligand and solvent on characters of ZnTe clusters. J. Mol. Struct. 938, 133–136.
G. Wiesenekker and E. J. Baerends (1991). Quadratic integration over the three-dimensional Brillouin zone. J. Phys. Condens. Matter 3, 6721–6742.
G. Kresse and J. Furthmüller (1996). Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50.
C. J. Pickard and R. J. Needs (2011). Ab initio random structure searching. J. Phys. Condens. Matter 23, 53201.
M. Franchini, P. H. T. Philipsen, and L. Visscher (2013). The becke fuzzy cells integration scheme in the amsterdam density functional program suite. J. Comput. Chem. 34, 1819–1827.
M. Franchini, P. H. T. Philipsen, E. Van Lenthe, and L. Visscher (2014). Accurate Coulomb potentials for periodic and molecular systems through density fitting. J. Chem. Theory Comput. 10, 1994–2004.
No Title, www.scm.com.
J. Eilers, E. Groeneveld, C. De Mello Donegá, and A. Meijerink (2012). Optical properties of Mn-doped ZnTe magic size nanocrystals. J. Phys. Chem. Lett. 3, 1663–1667.
H. H. Gullu, O. B. Surucu, M. Isik, M. Terlemezoglu, and M. Parlak (2020). Material and Si-based diode analyses of sputtered ZnTe thin films. J. Mater. Sci. Mater. Electron. 31, 11390–11397.
M. R. Gumaste and G. A. Kulkarni (2021). Dominant UV emission and n-type conductivity in manganese doped Zinc Telluride quantum dots. Phys. B Condens. Matter 615, 413076.
K. Megha, T. K. Mondal, Ghanty, and A. Banerjee (2021). Adsorption and Activation of CO2on Small-Sized Cu-Zr Bimetallic Clusters. J. Phys. Chem. A 125, 2558–2572.
Q. Gu, J. Huang, Y. Ma, K. Tang, H. Huang, Y. Hu, T. Zou, and L. Wang (2018). Properties of ZnTe Films Deposited with Different Substrate Temperature Using CSS Method. IOP Conf Ser Mater Sci Eng. https://doi.org/10.1088/1757-899X/362/1/012004.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ghulam Nabi, A., Rehman, A.u., Zainab, N. et al. First-Principles Study of Vibrational Modes and Raman Spectra in Mn-Doped ZnTe Nano- Clusters. J Clust Sci 34, 2309–2317 (2023). https://doi.org/10.1007/s10876-022-02380-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10876-022-02380-w