
Abstract
The development of carbon dots via a green synthesis approach from natural products is one of the most researched areas nowadays. Herein, we present the synthesis of Piper longum leaves-derived aqueous carbon dots (PLACDs) via the simplest ecofriendly hydrothermal carbonization method. The PLACDs exhibited excitation-dependent emission behavior with maximum emission at 450 nm at an excitation wavelength of 365 nm. The High-Resolution Transmission Electron Microscopy results showed a quasi-spherical shape with an average size of 4.121 nm. The sharp diffractions of X-ray diffraction revealed its nanocrystalline property. The Energy Dispersive X-ray spectra reflected the presence of carbon, nitrogen, and oxygen. The Fourier-Transform Infrared Spectroscopy disclosed the existence of –OH, –C=C, –C=O, and –C–O–C groups. The PLACDs presented excellent biocompatibility against B16F10 (melanoma) and SiHa (cervical carcinoma) cells lines with concentration-dependent in vitro bioimaging results. It also exhibited antiradical activity with the IC50 value of 0.499 mg/mL and 0.051 mg/mL against DPPH and ABTS.+, respectively. It showed Fe3+ sensing with a lower limit of detection of 0.673 μM. Further, the PLACDs displayed excellent bacterial biocompatibility, pH-dependent fluorescence property, fluorescent ink property, photostability, physical, chemical, and thermal stability.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Among various carbon families nanomaterials, such as three-dimensional graphite, two-dimensional graphene, and one-dimensional carbon nanotubes, carbon dots (CDs) or carbon quantum dots (CQDs) are recently discovered as superior zero-dimensional quasi-spherical functional carbon nanoparticles having the size of less than 10 nm [1]. The CDs are considered to be superior in comparison to semiconductor-derived quantum dots (QDs) due to their low toxicity, tunable emission property, high quantum yield, greater aqueous solubility, simple and versatile synthetic route, excellent physicochemical and photochemical stability, ecofriendly nature, cost-effective, excellent photoluminescence (PL), biocompatibility, good safety profile and wide opportunity for surface functionalization and surface passivation [2, 3]. Structurally they are nanocrystalline or amorphous materials having sp2 hybridized carbon with few expectations of sp3 carbon [1, 4]. Carbon, oxygen, and hydrogen are the major components of CDs; however, surface modification with heteroatoms such as sulfur, boron, or nitrogen can be done through suitable doping techniques for improving their optical properties [3]. CDs are extensively used in numerous fields such as bioimaging (in vitro cellular imaging and in vivo imaging) [1, 5], microbial cell imaging (bacteria, yeast and fungus) [6, 7], drug delivery [1, 8, 9], gene delivery [10], cancer theranostics [11], photodynamic therapy [12], sensing (drugs, small molecules, macromolecules, cations, anions, cells and bacteria) [13, 14], antibacterial agent [15], fluorescent ink [16], fingerprint detection [17], free radicals scavenging [18], catalysis (photocatalysis, electrocatalysis) [19, 20], and light-emitting diode [21]. The synthesis strategies of CDs are broadly classified into ‘‘top-down” (e.g., laser ablation, electrochemical exfoliation, arc discharge, oxidative acid treatment, etc.) and ‘‘bottom-up” methods (e.g., hydrothermal, thermal combustion, microwave, ultrasonic, etc.). During top-down processes, the breakdown of bulk carbon materials, such as graphite, carbon nanotubes, activated carbon, and carbon soot occurs. In contrast, in the bottom-up approach, the CDs are produced by “polymerization carbonization” approaches from molecular precursors such as biomass, carbohydrates, citrate, etc. [1, 2]. The top-down methods have the demerits of involvement of harsh conditions, require costly equipment and involve multiple steps [5]. In contrast, the bottom-up approach-based hydrothermal carbonization (HTC) offers advantages of simplicity, low cost, eco-friendly, easy scale-up ability, and ability to use a wide variety of accessible carbon precursors [16]. Again, the alteration of particle size or fluorescence property of the CDs can be achieved easily by changing volume of the sample, type of solvent, temperature, and duration of carbonization reaction. Further, the complete reaction in HTC occurs in a closed atmosphere of hydrothermal autoclave which avoid the direct exposure to the contained material. Moreover, the aqueous medium (aqueous extract) was used instead of costly and hazradous solvents in the HTC. Considering the aforementioned advantages, the CDs was prepared by HTC utilizing natural precursor and aqueous media.
Natural products, predominantly plants, have contributed a significant role in the healthcare of human beings from the primeval era to date [22, 23]. Plants are highly enriched in primary and secondary metabolites. The vast chemical diversities in plants represent it as a promising candidate for modern medicine [24]. CDs have been extensively synthesized by a green synthesis approach, utilizing green sources, such as leaves of Coriandrum sativum (Coriander, family: Apiaceae), Pseudo-stem of Musa acuminata (Banana, family: Musaceae), leaves of Bambusoideae (Bamboo, Family: Poaceae), Magnifera indica (Mango, family: Anacardiaceae), Solanum tuberosum (Potato, family: Solanaceae), Allium cepa (Onion, family: Liliaceae), and Ocimum sanctum (Holy Basil, family: Lamiaceae) [25]. The flowers, fruits, seeds, stems, and root parts of plant materials are highly enriched in diverse bioactive molecules. Due to the richness of carbon, oxygen, and nitrogen in natural sources, they act as a precursor for synthesizing CDs. During CDs preparation, the reaction kinetics, surface functionality, size, and quantum yield (QY) depend upon the available phytoconstituents in the biomass [16]. During hydrothermal treatment of natural sources, dehydration and carbonization occur with simultaneous in situ surface passivation under high temperature and pressure conditions [26]. Piper longum (P.longum, family- Piperaceae) is a well-known plant in Ayurveda, and commonly grows in evergreen forests of India. The whole plant, as well as various parts of the plant, are used for multiple ailments. Traditionally, the leaves are utilized for digestive disorders, stimulation of pancreatic lipase, eye ailment, cough, asthma, preventing infection, arresting milk secretion, constipation, etc. [27]. Compounds such as 2,2-Dimethoxybutane, β-Myrcene, 1,2-Benzenedicarboxylic acid, bis(2-ethylhexyl) ester are reported in the leaves of P.longum [28]. The essential oil from leaves contains α-Pinene, β-Pinene, limonene, linalool, γ-Elemene, caryophyllene, α-Muurolene, δ-cadinol, etc. [29]. Considering the wide chemical diversity of the leaves, they are selected as the precursor for the preparation of CDs.
The early diagnosis of tumors is the major issue that leads to cancer and causes serious health issues worldwide. Due to spatial resolution and excellent sensitivity, fluorescence imaging is one of the promising strategies in cellular imaging and target sensing [30]. Semiconductor-based QDs, Green fluorescent proteins (GFP), and fluorescent organic dyes were previously used as promising bioimaging probes due to their outstanding fluorescence features. However, poor photostability and mild fluorescence of GFPs make long-term imaging problematic, narrowing their extensive uses in biomedical fields. Most of the QDs possess poor aqueous solubility and high toxicity, making them inappropriate for live-cell or living body bioimaging [1, 31]. Therefore, exploring readily available, biocompatible novel fluorescent probes become crucial for in vitro and in vivo bioimaging applications. In this regard, CDs have been perceived as appropriate candidates as novel fluorescent probes for cellular bioimaging, especially target-specific imaging of the nucleus, cytoplasm, mitochondria, cellular membrane, microtubules, etc. [1]. They are economical, eco-friendly, photostable, less toxic, biocompatible, membrane permeable, possess weak interaction with proteins, are resistant to swelling, and are easily eliminated from the body [32]. Additionally, considering the excitation-dependent emission behavior of CDs, multicolor imaging is also possible [2]. Numerous research groups have widely investigated the bioimaging application of CDs against various cancer cells like melanoma (B16F10), cervical carcinoma (SiHa), breast cancer (MCF-7 and MDA-MB-231), cervical cancer (HeLa), lung cancer (A549), glioma (C6), osteoblast (MC3T3-E1), etc. [1, 31, 33, 34].
Free radicals are very unstable & extremely reactive molecules with an unpaired electron formed as a consequence of ATP production by the cellular mitochondria. Chief free radicals are the by-products of normal cellular metabolism, including reactive oxygen species (ROS) and reactive nitrogen species (RNS). Hydrogen peroxide (H2O2), superoxide anion, hydroxyl (OH−), and peroxyl (ROO−) radicals are the most common ROS, whereas peroxynitrite anion (ONOO−) and nitric oxide are the common RNS. These are involved in many biochemical reactions and denote an important part of aerobic life and metabolism. These radicals possess beneficial and toxic effects; thus play a dual role in biological systems [35,36,37]. They exert beneficial effects on immune functions and cellular responses at low or moderate levels. In contrast, they induce oxidative stress at high levels [37]. Oxidative stress cause several chronic and degenerative ailments like aging, autoimmune disorders, cancer, arthritis, ulcerative colitis, neurodegenerative and cardiovascular diseases [35, 37]. Antioxidant substances act as free radical scavengers that alter free radicals into waste by-products, which are ultimately removed from the body. They repair the free radicals-based damages by supplying electrons to these damaged cells, enhance the immune defense, and lower the risk of pathological conditions [37]. In this regard, CDs from leaves and stems of Tinospora cordifolia (Heart-leaved moonseed, family: Menispermaceae) [33, 34], leaves of Andrographis paniculata (Green chiretta, Family: Acanthaceae) [38], fruits of Citrus sinensis (Orange peels, family: Rutaceae) [39], Glycine max (Black soya beans, family: Fabaceae) [40], curcumin [41], proanthocyanidin [42], shell of Coffea arabica (Coffee bean, family: Rubiaceae) [43], essential oil of Thymus vulgaris (Garden Thyme, Family: Lamiaceae) [44] have been identified and well explored to be used as an antioxidant.
Iron is an essential micronutrient for living systems, including human beings, plants, and animals [45, 46]. It is an element of heme and chlorophyll. It is also a vital constituent of many enzymes, which plays an indispensable role in biological redox reactions [45]. Being an essential component of vital enzymes, such as cytochromes of the electron transport chain, it performs a variety of biological roles [46]. In the human body, the Fe3+ ion act as a key structural element or cofactor for multiple biochemical roles, including electron transfer, oxygen transport, cell respiration, enzymatic reaction, DNA, and hemoglobin synthesis. The Fe3+ ion should be within the optimal physiologic range for proper biological functions. Its shortage and additional quantity in a living system are the etiology for various disorders, such as renal failure, anemia, Alzheimer’s disease, heart disease, intelligence decline, Parkinson’s, and cancer [47,48,49]. Excess accumulation of iron in tissue induces iron-dependent oxidative stress [45]. Its excess deposition in skeletal muscle destructively interrupts muscle contractility [45]. A certain quantity of iron has a positive impact on the life activities of aquatic systems (plants and animals). However, with the growing economy, excess iron is accumulated in water, which can cause poisoning or death of aquatic systems [45]. Iron is an important micronutrient for topical plants whose shortage creates a global agricultural problem [46]. As an essential constituent of proteins and enzymes, it plays a substantial role in photosynthesis, chlorophyll synthesis, nitrogen fixation, respiration, DNA synthesis, and maintenance of chloroplast structure and function [46]. It also acts as an active cofactor of several enzymes that are essential for the synthesis of plant hormones, such as ethylene, lipoxygenase, and abscisic acid [46]. Iron deficiency is a general nutritional ailment in various crop plants, leading to low yields and decreased nutritional value [46]. In higher plants, stunted root development and interveinal chlorosis of young leaves indicate inadequate iron nutrition [46]. When iron accumulates above the threshold level, they become toxic. In such situations, excessive quantities of iron may be taken up by the plant, which encourages the formation of reactive oxygen-containing radicals, and ultimately destroys vital cellular organelles (e.g., cell membranes) by lipid peroxidation [45, 46]. Bronzing, acidity, and/or blackening of roots are indications of plants exposed to excess iron levels [46]. Thus, iron deficit harshly disturbs plant growth and development, and excess iron accumulation is toxic. Therefore, an optimal window for the iron concentration is needed to ensure smooth growth and development of the plant [46]. Therefore, it is very imperative to effectively and accurately analyze its level in biological and environmental samples (drinking water, soil) to prevent harmful impacts on biological systems. Currently, various methods including inductive coupled plasma-atomic emission spectroscopy, mass spectroscopy, atomic absorption spectrometry, UV–visible spectroscopy, fluorimetry, amperometry, ion chromatography, plasmon resonance Raleigh scattering spectroscopy, and electrochemical methods have been implemented for the quantitative and qualitative analysis of Fe3+ ion in various samples [47]. However, these techniques require sophisticated instruments, expensive chemicals, and tedious sample preparation procedures. Therefore, it is essential to develop cost-effective, easily available probes that can selectively sense Fe3+ accurately in the presence of other trace elements. Recently, CDs-based fluorescent probes have received substantial interest in identifying many metal ions, including Fe3+ ions with a high degree of selectivity and sensitivity [40, 41, 47, 48].
With the aim of exploring natural precursors for CDs synthesis, we report the single-step fabrication of highly luminescent eco-friendly carbon dots, i.e., Piper longum leaves aqueous carbon dots (PLACDs) by utilizing aqueous fresh leaves extract of Piper longum through a simple HTC technique. The in vitro cancer cell imaging, free radical scavenging, Fe3+ sensing activity, bacterial biocompatibility, fluorescent ink property, and stability of green synthesized carbon dots were explored in this research work. Our study encompasses the multidimensional application of PLACDs in pharmaceutical, biomedical, and bioengineering fields.
Materials and Method
Materials
Fresh leaves of P.longum were procured from Jayachandrapur village, district: Kendrapada, Odisha, in February 2020, and the voucher specimen (DM/PLL/PHYMEDLAB/2020) was deposited in the Phytomedicine Research Lab., Department of Pharmaceutical Engineering & Technology IIT (BHU), Varanasi. 3-(4, 5-dimethylthiazol- 2-yl)-2, 5-diphenyltetrazolium bromide (MTT), Dimethyl Sulfoxide (DMSO), Fetal bovine serum (FBS), Dulbecco's Modified Eagle's Medium (DMEM/F-12), 4′,6-diamidino-2-phenylindole (DAPI), Quinine sulfate, 1, 1-diphenyl-2-picrylhydrazyl (DPPH), ciprofloxacin, L-Ascorbic acid, 2,2'-azinobis-(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS.+), and 6-Hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid (Trolox) were obtained from Sigma-Aldrich (Sigma Aldrich, Germany). Potassium persulfate was purchased from Rankem chemicals (Haryana, India). Mueller–Hinton agar (MHA) media was obtained from Himedia Laboratories Pvt. Ltd. (Mumbai, India). B16F10 (melanoma) and SiHa (cervical carcinoma) cell lines were procured from the National Centre for Cell Science (NCCS), Pune, India. Multi-drug resistance pathogenic bacterial strains such as Klebsiella pneumoniae, Escherichia coli, Edwardsiella tarda, Staphylococcus aureus, Aeromonas hydrophila & Enterococcus faecalis were obtained from the Department of Microbiology, Institute of Medical Science, Banaras Hindu University. The Kcl, NH4Cl, CaCl2, NaCl, MgCl2, CuCl2 were obtained from Sisco Research Laboratories Pvt. Ltd. (Mumbai, India); BaCl2 and HgCl2 were purchased from Fisher Scientific Pvt. Ltd. (Mumbai, India); the AgNO3 received from Merck, NaAsO2, Na2HAsO4 obtained from SD Fine Chemical Ltd. (Mumbai, India), and FeCl3 obtained from Finar Chemicals Pvt. Ltd. (Gujarat, India). All the obtained chemicals are of analytical grade, hence directly used without any further purification or processing. Millipore water was used throughout the experiment whenever necessary.
Preparation of Extract
The P.longum leaves aqueous extract (PLLAE) was prepared by the cold maceration method. The partially grounded fresh leaves were dispersed in water at a proportion of 1:3.5 (w/v) and continuously agitated by an orbital shaker (REMI, RS 12 plus) to facilitate the extraction. After 24 h of continuous extraction, the aqueous extract was filtered and stored in a glass vial in the refrigerator until further use.
Preparation of CDs
The prepared aqueous extract (PLLAE) was utilized for the preparation of CDs via the hydrothermal carbonization (HTC) method. The PLLAE (100 ml) was transferred to a teflon-based cylindrical container contained in a stainless-steel hydrothermal autoclave (200 ml) and was subjected to a muffle furnace at 160 ºC for 8 h. After 8 h, the autoclave was allowed to cool naturally. A brown-colored liquid was obtained and centrifuged at 15,000 RPM for 10 min at 25 °C to remove unwanted coarse particles. The resultant supernatant was filtered through a 0.2 μm syringe filter (Pall-GelmanSupor Acrodisc®) to obtain clear Piper longum leaves aqueous carbon dots (PLACDs). The PLACDs was preserved in a glass vial wrapped with aluminum foil to avoid photobleaching and kept in a refrigerator at 4ºC until further use.
Instrumentation and Characterizations
UV–Vis absorption spectra of prepared PLACDs was recorded from 200-800 nm by UV–Visible spectrophotometer (Cary 60 UV Vis, Agilent, USA). The fluorescence spectra were recorded by a fluorescence spectrophotometer (Fluorolog-Horiba, HORIBA, Jobin Yvon, France) with a 1 nm slit width using the Xenon flash lamp as the light source. Particle surface morphology, Selected Area Electron Diffraction (SAED), and Energy Dispersive Analysis of X-rays (EDAX) spectra were investigated by High-Resolution Transmission Electron Microscopy (HRTEM) instrument (TECNAI G2 20 TWIN, FEI, USA) at 200 kV equipped with High Angle Annular Dark-Field (HAADF) detector. For HRTEM analysis, the PLACDs was diluted two times with Millipore water and dropped onto the carbon-coated copper grid (3.05 mm diameter, 400 Mesh, Ted Pella), and the excess sample on the grid was removed by soaking with clean filter paper, followed by drying to remove trace amount of residual solvent. The size of the PLACDs was measured by Image J software, and the average particle size with their distribution was traced from the measurement of 33 particles. The functional groups were identified by Fourier Transform Infrared Spectroscopy (FTIR) (Thermo Scientific Nicolet iS5 FTIR, USA) using KBr pellet within the wavenumber 4000–500 cm−1 with a minimum of 50 number scans and 4 cm−1 spectral resolution.
The drop cast technique was used for crystallinity analysis by Rigaku Miniflex 600 Desktop X-Ray Diffraction System (Tokyo, Japan) by means of CuKα radiation at 1.54 Å, 15 mA, and 40 kV. PLLAE and PLACDs were dropped separately on a clean glass slide (2.89 cm2 area) and dried at 50 °C for a whole night before XRD analysis. The films of samples were fitted to the sample holder and subjected to the XRD instrument. The d-spacing (interplane distance) was measured using Bragg's equation Eq. 1.
where, n is a positive integer (1), λ is the incident X-rays wavelength (λ = 1.54 Å), and θ is the angle between the surface of the materials and the incident rays.
The a-spacing (interatomic distance) was measured using Eq. 2.
where, “d” is the interplane distance, and h, k, and l are the Miller indices.
The fluorescent ink property of PLACDs was investigated by injecting it into an empty refill of ball pen and writing on filter paper (sd’s Zerohaze filter paper, Grade A, 125 mm). The written filter paper was visualized under visible and UV light (365 nm) to investigate the fluorescence ink property. The pH measurement was carried out using a digital pH meter (PC 700, Eutech Instrument, Singapore), which was formerly calibrated with standard buffer solutions of pH 4, pH 7 & pH10. Thermogravimetric analysis (TGA) was carried out by TGA instrument (Shimadzu, TGA-50, Kyoto, Japan) in a nitrogen atmosphere at a flow rate of 100 mL/min, using a platinum pan with a temperature rise at a rate of 10.00 °C/minute up to 800 °C. For TGA analysis, the PLACDs sample was dehydrated in a vacuum oven at 70 °C for a whole night, and the resultant dried sample of PLACDs was subjected to TGA analysis.
Measurement of Quantum Yield (QY)
The QY was calculated using Eq. 3 by measuring the fluorescence intensity of PLACDs with that of quinine sulfate (0.1 M H2SO4) as standard reference having QY 54% [6].
where, ‘QYPLACDs’ is the Quantum yield, subscript “PLACDs” represents carbon dots, and “QS” represents quinine sulfate. The “I” is the integrated fluorescent intensity of emission spectra, “A” is the absorbance at its fluorescence excitation wavelength, and “η” represents the refractive index of the solvent. The QY of PLACDs was assessed at its maximum excitation wavelength of 365 nm.
Stability Study of PLACDs
Photostability
The samples were exposed to UV radiation (365 nm) for 5 h, and a change in fluorescent property and intensity was observed.
Chemical Stability
The chemical integrity was identified by observing the changes in FTIR and UV spectra of the of PLACDs sample stored in a refrigerator at an interval of six months up to 12 months.
Physical Stability
Prepared PLACDs were stored for a prolonged period in the refrigerator and checked for changes in color, appearance, signs of agglomeration, precipitation, and sedimentation.
pH-Dependent Fluorescence Stability
It was evaluated by exposing the PLACDs to various aqueous pH solutions (prepared by mixing different volumes of 0.1 N HCl or NaOH) and evaluating the variation in fluorescence intensities.
Thermal Stability
TGA analysis was performed to investigate the thermal stability of PLACDs by noticing the material’s weight loss with respect to temperature.
In vitro cytotoxicity and bioimaging.
Cell lines Procurement and Maintenance
The B16F10 (melanoma) and SiHa (cervical carcinoma) cell lines were procured from the National Centre for Cell Science (NCCS), Pune, India. Cell lines were cultured in a DMEM/F-12 medium containing 50 unit/mL streptomycin and penicillin with 10% fetal bovine serum (FBS) and incubated in a CO2 incubator maintained with 5% CO2 at 37 °C.
In vitro Cytotoxicity Assay
MTT assay was performed to evaluate the cytotoxic potential of aqueous extract (PLLAE) and prepared carbon dots (PLACDs) on B16F10 and SiHa cells. The assay includes the mitochondrial enzymatic alteration of formazan crystals from MTT. The cancerous cells (1 × 106/well) were seeded in a 96 microtiter well plate in DMEM/F-12 with 10% FBS solution, 50 unit/mL penicillin & streptomycin and incubated with 5% CO2 atmosphere at 37 °C for 24 h to allow the adherence of cells and grow up to 60 to 70% of confluency. After incubation, the medium was removed & further incubated with fresh media containing different concentrations (0–300 µg/mL) of PLLAE and PLACDs for another 24 h. Then, the media was removed, the samples were replaced with 200 μL of serum-free media containing 20 μL MTT solution (5 mg/mL), and cells were incubated for 4 h at 37 °C to allow the formation of insoluble formazan crystals by viable cells. The MTT-containing media was removed, and the DMSO (150 μL) was added and incubated for 15 min at 37 °C to solubilize insoluble formazan crystals produced by viable cells. Then the absorbance was taken by a microtiter plate reader (Bio-Rad Laboratories, Munchen, Germany) at 570 nm. The cell viability percentage was calculated using Eq. 4.
where, \({A}_{s}\) and \({A}_{C}\) are the absorbance with sample (PLLAE or PLACDs) and absorbance of the control (no treatment), respectively.
In vitro Cancer Cell Imaging and Uptake Study
The in vitro cellular imaging potential of PLACDs was examined by fluorescence microscopy [16]. The cells (1 × 105 cells/well) were seeded in a six-well plate in DMEM/F-12 media containing 10% FBS, streptomycin & penicillin (50units/mL) and incubated in a humidified incubator with 5% CO2 at 37 °C for 24 h. Then the staining was achieved by replacing media with PLACDs (0-75 µg/mL) and incubating for another 24 h. After staining, the cells were fixed with paraformaldehyde (4%), washed three times with PBS followed by nuclear counterstaining with DAPI, and mounted on the platform of the fluorescence microscope (EVOS® FL cell imaging system, Thermo Fisher Scientific, USA) for obtaining images.
Free Radical (DPPH and ABTS..+) Scavenging Activity
The scavenging activity of PLLAE and PLACDs was evaluated by DPPH and ABTS.+ assay method [18, 50,51,52].
DPPH Assay
A 100 μM fresh solution of DPPH was prepared in methanol in a dark environment. The PLLAE or PLACDs was diluted suitably with water to produce each stock solution of 3.7 mg/mL concentration. Then, different volumes of stock solution of PLLAE and PLACDs were taken in multiple test tubes, and the volume was adjusted up to one milliliter with water to obtain a series of concentrations of samples. Accurately 2 mL of methanolic DPPH solution was added to the 1 mL of each diluted sample (PLLAE or PLACDs), in which the final concentrations of samples in total 3 mL of the solution was ranged from 0 to 0.861 mg/mL. The samples were incubated in a dark chamber for 30 min. Ascorbic acid was chosen as a positive control. Different volumes of the aqueous solution of ascorbic acid (100 μg/mL) were diluted with water up to 1 mL to produce a series of concentrations. Then, 2 mL of methanolic DPPH solution was added to 1 mL of diluted ascorbic acid. The concentrations of ascorbic acid in the final 3 mL solution ranged from 0 to 8 μg/mL. The same above-cited procedure was employed for ascorbic acid. The results of DPPH scavenging activity were represented in percentage (%) and calculated by Eq. 5.
where, Abscontrol is the absorbance of the control (DPPH radicals in the absence of any sample or standard) & Abssample or standard is the absorbance of DPPH radicals with sample or standard at 517 nm. The IC50 value (in μg/ml) was estimated by calculating the concentration of treatments that cause 50% of scavenging.
ABTS+ Assay/Trolox Equivalent Antioxidant Capacity
Like DPPH radical, the ABTS.+ does not exist readily, hence generated chemically. The ABTS.+ radical was produced by mixing 7 mM ABTS aqueous solution with 2.45 mM potassium persulfate aqueous solution at a ratio of 1:1 v/v and allowing it to stand for 16 h in the dark until the completion of the reaction at which the absorbance was stable. The ABTS.+ solution was further diluted with water to an absorbance of 0.740 ± 0.04 at 734 nm. The PLLAE or PLACDs was diluted suitably with water to produce each stock solution of 3.7 mg/mL concentration. Then, different volumes of stock solution of PLLAE and PLACDs were taken in test tubes, and the volumes were adjusted up to one milliliter with water to obtain a series of concentrations of samples. Accurately 3 mL of aqueous ABTS.+ solution was added to the 1 mL of each diluted sample (PLLAE or PLACDs), in which the final concentrations of samples in total 3 mL of the solution was ranged from 0–0.647 mg/mL. The samples were incubated in a dark chamber for 5 min, and immediately the absorbances were taken at 734 nm. Trolox (water-soluble analogous to vitamin E) was used as a positive control. Different volumes of the aqueous solution of Trolox (100 μg/mL) were diluted with water up to 1 mL to obtain different concentrations. Then, 3 mL of aqueous ABTS.+ solution was added with 1 mL of diluted Trolox. The concentrations of Trolox in the final 4 mL solution ranged from 0–7.5 μg/mL. The results of ABTS.+ scavenging activity were represented in percentage (%) and calculated by Eq. 6.
where, Abscontrol is the absorbance of the control (ABTS.+ without any sample or standard) & Abssample or standard is the absorbance of ABTS.+ with sample or standard at 734 nm. The IC50 value (in μg/ml) was estimated by calculating the concentration of treatments that cause 50% of scavenging of ABTS.+ radical.
Metal Ions Sensing
The aqueous solution of metallic ions (Mg2+, Ba2+, Fe3+, Ag3+, Hg2+, Cu2, As3+, As5+, NH4+, Ca2+, K+, and Na+) were prepared from their respective salt form. Accurately 2 mL of each metal solution was added with 2 mL of diluted PLACDs (0.837 mg/mL) in order to achieve 250 µM each metal ion & 0.418 mg/mL of PLACDs in final volume (i.e.,4 mL). Control sample was prepared by addition of 2 mL of diluted PLACDs (0.837 mg/mL) with 2 mL of water to achieve final concentration of 0.418 mg/mL. Further, the PLACDs-metallic solutions were incubated at room temperature for 15 min, and their fluorescence intensities were measured at the maximum excitation wavelength (365 nm) with 1 nm slit width. The metallic ion that greatly affects/quenches the fluorescence intensity was identified and used for the sensitivity analysis. The fluorescence titration method estimated the sensitivity of the prepared PLACDs towards selected metal ions (Fe3+). About 2 ml of diluted PLACDs (0.837 mg/mL) was mixed with 2 mL of Fe3+ solution to obtain 0-1000 μM Fe3+concentrations in total 4 mL of solution. All solution mixtures were incubated for 15 min, and fluorescent measurement was performed at 365 nm. The fluorescence quenching efficiency of PLACDs was calculated by Stern–Volmer relation as in Eq. 7 [49].
where, F0 and F are the fluorescence intensities of PLACDs in the absence and presence of metal ions, respectively, Ksv is the Stern–Volmer quenching constant, and Q is the metal ion concentration.
The limit of detection (LOD) of the prepared PLACDs on selected metal ions in water was determined using Eq. 8 [49].
where, σ is the standard deviation of y-intercept (F0/F values), and s is the slope of the linear line.
Bacterial Biocompatibility
It was studied on pathogenic bacterial strains by the conventional disc diffusion method [53]. The Gram-negative bacterial strains (Klebsiella pneumoniae, Edwardsiella tarda, Escherichia coli & Aeromonas hydrophila) and Gram-positive strains (Staphylococcus aureus & Enterococcus faecalis) were used for the study. The bacterial suspensions of 0.5 McFarland optical density were prepared in isotonic NaCl solution and spread over the surface of semi-solid sterile Mueller–Hinton agar (MHA) and incubated for 30 min at 37 °C. After initial incubation, ten microliters of each sample, i.e., PLLAE (14.8 mg/mL) and PLACDs (14.8 mg/mL), were dropped on the surface of sterile filter paper discs (6 mm diameter) placed on the surface of MHA media and further incubated at 37 °C for 24 h. Ciprofloxacin (20 μg/disc) aqueous solution was used as a positive control due to its broad spectrum of activity. Another disc loaded with sterile water was used as a negative control. The zone of inhibition around samples was measured by a scale.
Results
Synthesis of PLACDs
The present research aims to develop highly fluorescent eco-friendly CDs from plant sources via a green synthesis approach. The synthetic protocol of PLACDs was schematically presented in Fig.S1a.
Optical Properties
The optical possessions of PLACDs were explored by UV–Vis spectroscopy and fluorimetry. The UV–Vis spectra of PLACDs and its precursor (PLLAE) are shown in Fig. 1a. PLACDs showed two broad characteristic absorption peaks at 270 nm and 320 nm corresponding to π–π* transition of C = C bonds and n- π* transition of C = O bond, respectively [49, 54]. The PLACDs showed two excitation peaks (Fig. 1b) at 301 nm and 365 nm, and the corresponding maximum emission peak wavelengths were found to be 445 nm and 450 nm (Fig. 1c), respectively. Out of two excitation maxima, the PLACDs showed maximum fluorescence intensity for excitation at 365 nm (Fig. 1c). To further explore the optical properties, the fluorescent spectra of PLACDs were obtained at multiple excitation wavelengths (Fig. 1d). The CDs exhibited maximum fluorescence emission at 450 nm at an excitation of 365 nm (Fig. 1d). Further, the 2-D contour plot also revealed the maximum fluorescence intensity (5.328 × 104 a.u.) of PLACDs (431.06 µg/mL) at emission maxima (450 nm) with an excitation of 365 nm (Fig. 1e). The fluorescence intensity was further observed for various concentrations of PLACDs. A gradual increase in fluorescence intensity was observed from concentration 49.16 to 431.06 µg/mL (Fig. 1f). The calculated QY of PLACDs at an excitation wavelength of 365 nm was found to be 3.32% against standard reference quinine sulfate. Our result is in accordance with the previous reports on the QY of CDs synthesized by the green chemistry approach [33, 34, 55,56,57]. The PLACDs was explored as fluorescent ink for writing and drawing purposes.

Optical properties of PLACDs a UV–Vis spectra of PLACDs and PLLAE, b Excitation spectra of the synthesized PLACDs, c Emission spectra of the synthesized PLACDs at excitation wavelengths 301 nm and 365 nm, d Excitation dependent emission spectra of PLACDs, e Excitation-emission contour plot of PLACDs, and f Fluorescence spectra of different concentrations of PLACDs at an excitation wavelength of 365 nm
HRTEM, Size Distribution, and EADX
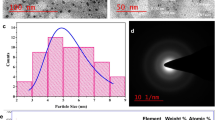
Figure 2a-e shows the HRTEM photomicrographs, SAED pattern, particle size distribution, and EDAX results of PLACDs. The PLACDs particles dispersed on the Cupper grid were found to be nearly quasi-spherical in shape (Fig. 2a, Fig. 2b, and Fig. 2c). The SAED analysis of selected PLACDs particles on the HRTEM grid showed diffused rings, signifying its amorphous nature (Inset Fig. 2c). The diameter of 33 dots measured by ImageJ was found to be ranging from 2 to 7 nm, with an average diameter of 4.121 nm (Fig. 2b, d). The EDAX spectra reflected the presence of carbon, oxygen, nitrogen, phosphorus, and sulfur with a higher percentage of carbon (75.73%), followed by oxygen (17.61%), nitrogen (5.94%), and other elements on the surface of PLACDs (Fig. 2e).

HRTEM, SAED, Particle size distribution, and EDAX analysis of PLACDs a HRTEM photomicrograph of PLACDs at 50 nm scale, b HRTEM photomicrograph of PLACDs at 50 nm scale analyzed through ImageJ software for particle size analysis c HRTEM photomicrograph of PLACDs at 100 nm scale with inset image showing the SAED pattern, d Particle size distribution of PLACDs e EDAX spectra of PLACDs with inset data reflecting the elemental composition
Fourier Transform Infrared Spectroscopy (FTIR)
The surface functionality of PLACDs and PLLAE were investigated by FTIR spectroscopy (Fig. S1b). A broad peak at 3450 cm−1 was ascribed to alcoholic or phenolic O–H stretching. A sharp peak at 1645 cm−1 was due to amidic C=O stretching. Two weak peaks at 1406 cm−1 and 1065 cm−1 are ascribed to aromatic C=C stretching or O–H bending of carboxylic acid and C–O–C stretching, respectively. Similar spectral results were also obtained with PLLAE (Fig. S1b).
XRD
The PLLAE showed sharp and strong diffraction peaks at 2θ of 28.038° and very weak diffractions at 40.22° and 58.181° (Fig.S1c). The XRD pattern of PLACDs (Fig.S1c) showed an intense peak at 2θ = 28.037° and weak diffraction peaks at 2θ = 40.18°, 50.023°, and 58.88°. The peaks at 50.023° and 58.88° are ascribed to C(102) and C(103) planes of graphitic carbon, respectively [58]. The calculated inter-plane distance (d) of PLACDs is approximately 0.182 nm and 0.159 nm for the refraction peaks of C(102) and C(103), respectively. The calculated inter-atomic distance (a) corresponding to C(102) and C(103) was 0.406 nm and 0.502 nm, respectively.
Fluorescent Ink Property
The photograph of an empty refill of ball pen loaded with aqueous PLACDs solution as the ink is shown in Fig.S1d. Fig.S1e clearly displays the absence of any write-up or drawings in visible light. In contrast, the Odia language word “maa” in the middle and border drawings on filter paper were clearly visualized under long UV light (Fig.S1f).
Stability Studies
Photostability
The PLACDs were exposed to UV irradiation at 365 nm for 5 h and examined intermittently for possible fluorescent behavior and intensity changes. After five hours of constant UV exposure, no photobleaching or such changes in emission were noticed while observed by the naked eye (Fig.S2a). The data obtained from the fluorescent spectrophotometer reflects a consistent spectral pattern with no significant alteration of fluorescent intensity (Fig.S2b).
Chemical Stability
The FTIR and UV spectral data of PLACDs are shown in (Fig.S2c and Fig.S2d). Consistent spectral results were obtained throughout the study.
Physical Stability
Various organoleptic properties and physical parameters were monitored during the long-term storage of PLACDs. No change in color or appearance was observed at an interval of 6 months up to 12 months (Fig. S2e).
pH-Dependent Fluorescence Stability of PLACDs
The fluorescence spectra (Fig.S2f) were recorded by exposing 90µL of PLACDs (14.8 mg/mL) to the 3 mL aqueous solution of different pH. With the increase of pH from 1–9, a steady elevation in the fluorescent intensity was observed, with maximum intensity (7.325 × 105) at pH 9, after which a decrease in intensity was observed.
Thermal Stability
The weight loss in a controlled atmosphere with respect to elevation in temperature was accessed by the TGA experiment (Fig.S2g). The PLLAE did not show any degradation up to 102.34 °C, after which it showed 3-step degradations with a continuous weight loss of 38.76% between 102. 34 °C and 320 °C, 14.12% between 320 °C and 501.45 °C, and 12.84% between 501.45 °C and 800 °C. The PLACDs showed four-step degradations with continuous weight loss of 4.25% between 27.64 °C and 163.93 °C, 14.26% between 163.93 °C and 343.14 °C, 13.45% between 343.14 °C and 501.17 °C, and 13.75% between 501.17 °C and 800 °C were observed.
In Vitro Cytotoxicity Studies and Fluorescent Imaging
The cytotoxic potential of PLLAE and PLACDs was evaluated by MTT assay on B16F10 and SiHa cell lines. The percentage cell viability corresponding to various concentrations of PLLAE or PLACDs was presented in Fig. 3a, b. The PLACDs exhibited slightly higher cytotoxicity in the tested cell lines compared to its precursor PLLAE. Considering water as a dispersion medium and excellent fluorescent properties of the prepared PLACDs, it was examined for in vitro live-cell imaging applications. The in vitro cellular imaging was performed on B16F10 and SiHa cell lines using PLACDs as a fluorescent probe below 80 μg/mL concentration. In both cell lines, the PLACDs treated cells showed green color fluorescence while excited under UV irradiation. In contrast, the PLACDs untreated cells did not display any fluorescence (Fig. 3c, d). Concentration-dependent fluorescence property was obtained with PLACDs from 0 to 75 μg/mL. The PLACDs was found to be successfully reached the cytoplasmic region, confirmed by nuclear fluorescence by DAPI.


Cell viability (%) and In vitro bioimaging of cancer cells a Cell viability of melanoma (B16F10) cells after 24 h of incubation with different concentrations of PLLAE and PLACDs, b Cell viability of cervical cancer (SiHa) cells after 24 h incubation with different concentrations of PLLAE and PLACDs, c Bioimaging of melanoma (B16F10) cell lines and d Bioimaging of cervical cancer (SiHa) cell lines under bright field, blue field (DAPI staining), green field (PLACDs treated) and merged micrographs of DAPI and PLACDs after 24 h of incubation
Free Radical Scavenging Activity
Concentration-dependent DPPH scavenging activities were found with extract (PLLAE) and PLACDs with gradual color change from deep purple to light yellow (Fig. 4a, b). This is further confirmed by the decline in the absorbance upon a gradual increase in the concentrations of PLLAE or PLACDs from 0 to 0.861 mg/mL (Fig. 4c, d). The linearity was obtained within the PLLAE concentration of 0.123 to 0.861 mg/mL with the regression equation y = 73.428x + 7.607 and correlation coefficient (R2) value of 0.982 (Fig. 4e). The IC50 value of PLLAE from the equation was found to be 0.577 mg/mL. Likewise, the linearity found within the PLACDs concentration of 0.049 to 0.762 mg/mL with the obtained regression equation y = 112.43x-6.117, R2 value of 0.997 and IC50 value 0.499 mg/mL (Fig. 4f and Inset Fig. 4f). The IC50 value of the ascorbic acid as a positive control was found to be 6.727 μg/mL (Fig. 4g) calculated from the regression equation y = 6.745x + 4.619 and R2 value of 0.998.

DPPH radical scavenging assay of PLLAE and PLACDs a Photograph of DPPH solution exposed to different concentrations of PLLAE, b Photograph of DPPH solution exposed to different concentrations of PLACDs, c UV absorption spectra of DPPH radical upon a gradual increase in PLLAE concentration from 0 to 0.861 mg/mL d UV absorption spectra of DPPH radical upon a gradual increase of PLACDs concentration from 0 to 0.861 mg/mL e Calibration curve of percentage scavenging of DPPH radicals v/s PLLAE concentrations f Calibration curve of percentage scavenging of DPPH radicals v/s PLACDs concentrations with inset graph representing the linearity region, and g Calibration curve of percentage scavenging of DPPH radical v/s different concentrations of ascorbic acid
Concentration-dependent ABTS˙+ scavenging activities were obtained with extract (PLLAE) and PLACDs with a gradual change in color from a blue-green to a colorless solution (Fig. 5a and Fig. 5b). Further, it was confirmed from the decline in the absorbance upon a gradual rise in the concentrations of PLLAE or PLACDs from 0 to 0.647 mg/mL (Fig. 5c, d). The linearity was obtained within the PLLAE concentration of 0.037 to 0.092 mg/mL with the linear equation Y = 631.595x + 12.201 and correlation coefficient (R2) value of 0.996 (Fig. 5e and inset Fig. 5e). The IC50 value of PLLAE from the linear equation was found to be 0.059 mg/mL. Similarly, the linearity was found within the PLACDs concentration of 0.037 to 0.092 mg/mL with the linear equation Y = 618.99x + 17.87, R2 value of 0.994 and IC50 value 0.051 mg/mL (Fig. 5f and Inset Fig. 5f). The dose-dependent scavenging results were obtained up to the concentration of 0.203 mg/mL after which a steady-state was attained. The IC50 value of the Trolox estimated from the regression equation Y = 14.674x—8.55 (R2 = 0.996) was found to be 3.99 μg/mL (Fig. 5g).

ABTS.+ scavenging assay of PLLAE and PLACDs a Photograph of ABTS.+ solution exposed to different concentration of PLLAE, b Photograph of ABTS.+ solution exposed to different concentration of PLACDs, c UV absorption spectra of ABTS.+ radical upon a gradual increase in the PLLAE concentration from 0 to 0.647 mg/mL d UV absorption spectra of ABTS.+ upon a gradual increase in the PLACDs concentration from 0 to 0.647 mg/mL e Calibration curve of percentage scavenging of ABTS.+ radicals v/s PLLAE concentrations with inset graph representing the linearity region f Calibration curve of percentage scavenging of ABTS.+ v/s PLACDs concentrations with inset graph representing the linearity region, and g Calibration curve of percentage ABTS..+ scavenging v/s Trolox concentrations
Sensing of Metal Ion
The selectivity of PLACDs towards multiple metal cations was evaluated by studying the quenching of the fluorescent intensity upon the addition of metal ions. Changes in the fluorescent intensities of PLACDs (0.418 mg/mL) in the presence of 250 μM of various metal ions and in the absence of ions were shown in Fig. 6a, b. The results display a substantial quenching of fluorescence intensity in the presence of Fe3+, reflecting higher selectivity of PLACDs towards the sensing of Fe3+.

Metal ion sensing property of PLACDs a Fluorescence spectra of PLACDs in the presence of different metal ions (250 µM), b Relative fluorescence intensity of PLACDs in the presence of 250 µM different metal ions, c fluorescence spectra of PLACDs in the presence of different concentrations (0–1000 µM) of Fe3+, d Fluorescence intensity of PLACDs v/s Fe3+ concentrations from 0 to 1000 μM (Inset image reflecting linearity region), and e calibration curve of relative fluorescence intensity of PLACDs (F0/F) v/s Fe.3+ concentration from 0 to 1000 μM
The sensitivity of PLACDs towards Fe3+ ions was explored by fluorescent titration assay [49]. The decrease in the fluorescent intensity of PLACDs (0.418 mg/mL) was observed by incubating with various concentrations (10–1000 μM) of Fe3+ ion. A steady decrease in the fluorescence intensity was noticed with a rise in the Fe3+ concentration (Fig. 6c, d). A linear relationship exists within 0–100 μM concentration with the corresponding linear equation y = 30,670.872–66.296x (Inset Fig. 6d). The value of R2 (correlation coefficient) was found to be 0.998, describing strong sensitivity towards Fe3+ ions.
Stern–Volmer relation was utilized to evaluate the quenching efficiency of the PLACDs in terms of LOD [49]. The value of F0/F was plotted against the concentration range (0–100 μM) of Fe3+ ions (Fig. 6e). The corresponding linear regression equation was F0/F = 0.003x + 1.072 with a coefficient of correlation of 0.998 and y-intercept 1.072. The value of slope or Ksv (Stern–Volmer quenching constant) was found to be 0.003. The LOD value was found to be 0.673 μM, which is very low, suggesting the higher sensitivity of PLACDs.
Bacterial Biocompatibility
The bacterial biocompatibility of PLACDs in terms of zone of inhibition was studied by disc diffusion assay (Fig.S3a-f). The antibiotic ciprofloxacin showed a larger zone of inhibition in all strains, reflecting the susceptibility towards the experimental microorganisms. The tested samples, i.e., PLLAE and PLACDs, showed no zone of inhibition which is similar to that of the negative control (water), reflecting the bacterial biocompatibility. Thus, the biocompatible fluorescent PLACDs may be used for live bacterial imaging.
Discussions
The leaves of P.longum are highly enriched in C, N, and O atoms, thereby acting as a precursor for CDs synthesis. Functional groups of multiple phytoconstituents in aqueous extract (PLLAE) act as passivating agents for CDs synthesis. As a result, we have synthesized CDs (PLACDs) from PLLAE via one-step hydrothermal carbonization. The hydrothermal method was preferred due to its simplicity in synthesis, cost-effective, eco-friendly, and non-toxic characteristics [5]. After eight hours of hydrothermal digestion, a light brown colored product with a characteristic odor was found (Fig.S1a), suggesting the effective carbonization and synthesis of CDs. The emission of bluish-green fluorescence (Fig.S1a) under UV lamp (at 365 nm) further confirmed the synthesis of carbon dots (PLACDs).
The optical possessions of CDs depend upon their size, radiative recombination of excitons, arrangement of emissive trap sites on the surface, and free zigzag sites [58]. The UV–vis spectrum of PLACDs resembles the spectrum of PLLEE (Fig. 1a), representing the retention of chemical integrity of phytoconstituents of PLLEE in PLECDs. The fluorescent property of CDs depends upon surface functionality and size [19, 58, 59]. A fluctuation in fluorescence intensity and double emission peaks were observed between 280 to 320 nm (Fig. 1d). A steady increment in the fluorescent intensity (Fig. 1d) was noticed between excitation wavelengths of 320 nm to 365 nm without significant shifting of emission spectra, possibly due to π* to π transition of graphitic carbon [33, 34, 60]. However, the decrease in fluorescent intensity with the Bathochromic shift was observed from excitation wavelength between 365–400 nm (Fig. 1d). Such excitation-dependent emission property of CD was also reported previously [33, 34, 49, 60]. The fluorescent nature of the PLACDs is attributed to surface defects and the quantum confinement effect. The existence of multiple surface defects brings about the energy centers, which provide multiple emissions, and the quantum confinement effect due to their miniature particle size confers to good stability [3]. The broad range of fluorescent spectra reflects the potential of PLACDs as a probe for bioimaging applications.
HRTEM analysis provides an idea about the surface morphology of the nanomaterials [61]. The PLACDs was found to be quasi-spherical shape (Fig. 2a–c) with an average diameter of 4.121 nm (Fig. 2b, d). The maximum percentage of carbon in the EDAX result (Fig. 2e) represents the carbonaceous nature of PLACDs. The EDAX spectra of Cu are due to its presence in the TEM grid.
The characteristic infrared spectral signature of PLACDs is similar to that of PLLAE (Fig. S1b), suggesting the retention of functional groups and phytochemical composition of PLLAE in the PLACDs. The data is well consistent with the similar UV–Vis spectral data of PLACDs and its precursor PLLAE (Fig. 1a).
The XRD pattern (Fig.S1c) of PLLAE showed a combination of strong diffraction peaks (2θ = 28.038°) and very weak diffractions (2θ = 40.22° and 58.181°), which are mainly ascribed to the presence of numerous crystalline phytoconstituents in the plant extract. The XRD result of PLACDs (Fig.S1c) demonstrated a nearly similar diffraction pattern to that of its precursor (PLLAE), representing the retention of the phytochemical composition of PLLAE in the prepared PLACDs. The data is in accordance with the FTIR and UV results. The prepared PLACDs may be a mixture of PLACDs and PLLAE. The highly intense sharp diffraction peak in PLACDs at 2θ = 28.03° and weak peaks at 2θ = 40.18° and 58.88° may be contributed by the aqueous extract (PLLAE). The peaks at 50.02° and 58.88° of PLACDs are ascribed to C(102) and C(103) planes of graphitic carbon, respectively [58]. The XRD data of PLACDs is in good agreement with the diffractogram of CDs prepared via the hydrothermal treatment of Solanum tuberosum (family: Solanaceae) [58]. Structurally CDs are either nanocrystalline or amorphous nanomaterials [1]. Mostly, the XRD pattern of many CDs was reported as halo diffraction patterns [33, 34, 48]. Besides the intense peaks contributed by PLLAE, the PLACDs of its own showed very less intense diffraction peaks, suggesting a nearly amorphous nature. This is further confirmed by the diffused rings pattern (inset Fig. 2c) of the SAED analysis of one of the selected PLACDs particle.
The CDs can be used as an alternative to traditional inks due to their excellent fluorescence property, durability, easy washing ability, and biocompatibility characteristics. The word & drawing written by the PLACDs was found to be invisible in visible light (Fig.S1e); however, they emitted fluorescence and visualized while observed under UV light (Fig.S1f), thus may be used as fluorescent ink for security purposes for the writing of confidential matters. During the write-up, the flow of the fluorescent ink through the refill of the ball pen was very good. However, in terms of performance, there is still a certain gap in using the aqueous solution of PLACDs as the ink directly due to its low viscosity, which may leak out from the refill.
For bioimaging application, the fluorescent probe must possess constant photostability during a long-term examination. The constant fluorescence property (Fig.S2a & Fig S2b) confirms the good photostability of PLACDs. The constant fluorescence intensity of UV exposed PLACDs reflects its worthiness as a fluorescent probe for bioimaging applications. From the chemical stability study, no significant changes in the spectra of FTIR and UV were observed while analyzed at 6 and 12 months, indicating the long-term chemical integrity of the PLACDs (Fig.S2c and Fig.S2d). As per Stokes' law, the physical stability of nanodispersion depends upon particle size, the density of the dispersed phase and dispersion medium, and the viscosity of the medium [62]. Decreasing particle size, balancing density of particle and medium, or increasing the viscosity of medium produce stable colloidal suspension with minimum sedimentation. The prepared PLACDs remain stable for 12 months without any sign of agglomeration, sedimentation, or floating precipitates (Fig. S2e). The overall results reflected the good physical stability of PLACDs. The pH-dependent fluorescent intensity changes (Fig. S2f) were assigned to modification in the surface state due to the ionization of functional groups [33, 34, 54]. The fluorescence intensity of studied PLACDs was found to be stable in a wide pH range (pH 3–11), reflecting its probable biological applications in the presence of various physiological media of different pHs.
TGA analysis represents the thermal stability, composition, and surface functionality of the analyte [58, 63]. The PLLAE did not show any weight loss up to 102.34 °C (Fig. S2g), representing its thermal stability up to that temperature, after which it exhibited 3-step degradations due to degradation of contained phytomolecules. In contrast, the PLACDs showed four steps degradation with continuous weight loss up to 800 °C (Fig. S2g), reflecting the alteration of the thermal behavior of the contained molecules during hydrothermal treatment. The initial slight weight loss (4.25%) is ascribed to the degradation of weakly bonded chemical groups bonded by weak hydrogen bonding [58]. At a later step, gradual weight loss of 14. 26%, 13.45%, and 13.75% until 800 ºC were assigned to degradation of functional groups that are strongly anchored to the surface of PLACDs via covalent linkages [63]. This confirms the existence of varieties of chemical groups on the surface of PLACDs.
Cytotoxicity of a sample plays a vital role in determining its suitability as a fluorescent probe for bioimaging applications. A slightly higher cytotoxic potential of PLACDs (Fig. 3a, b) may be due to its nanostructure, which causes enhanced penetration into the cells compared to PLLAE. The percentage cell viability of both cancer cells at concentrations lower than 80 μg/mL was found to be nearer to or more than 80% after 24 h of incubation. This result showed the low cytotoxicity of the PLACDs below this concentration and inferred its probable application for in vitro cancer cell imaging. Ideally, fluorescent probes for in vitro viable cell imaging should be water-soluble, photostable, highly fluorescent, chemically inert, and biocompatible [34]. Utmost the presently used fluorescent dyes and semiconductor-based quantum dots are unstable, poorly water-soluble, and highly toxic, thereby making them inappropriate for in vitro cellular imaging. In this regard, CDs have been perceived as an appropriate agent for bioimaging [1]. The PLACDs showed low cytotoxicity towards both cell lines (Fig. 3a, b) and good in vitro bioimaging results (Fig. 3c, d). Considering the good aqueous solubility, outstanding fluorescence behavior at lower concentrations, and cellular biocompatibility, the PLACDs showed its excellency as a fluorescent probe for cancer cell imaging.
DPPH and ABTS.+ radicals have representative absorption maxima at 517 nm and 734 nm, respectively, which vanishes after their scavenging by an antiradical/ antioxidant substance. Thus, the reduction of DPPH or ABTS.+ can be checked by evaluating the decrease in its absorbance at their respective absorption maxima (λmax) during the reaction with antioxidant substances. These methods are easy, precise, and inexpensive and are widely used for the screening of antiradical compounds [51]. In the presence of antioxidant substances, the purple-colored nitrogen-containing free radical changed to yellow color. After accepting H, the DPPH radical converted to a stable neutral DPPH-H complex [18]. The DPPH scavenging activity of CDs was also previously reported by some research groups [18, 33, 34, 38, 54, 64]. The PLACDs showed excellent DPPH scavenging potential (Fig. 4d, f). The obtained IC50 of PLACDs (0.499 mg/mL) is low as compared to the PLLAE (0.577 mg/mL). This might be due to the availability of unpaired electrons, nanostructure, high surface-to-volume ratio, and presence of surface defects of PLACDs, making them more sensitive to scavenge free radicals at a lower concentration [65]. Such higher DPPH radical scavenging potential of CDs compared to its precursor (extract) was also previously reported by our research groups [33, 34]. The ABTS.+ assay method is based on the scavenging of ABTS˙+ radical by absorbing electrons or protons from antioxidants [52]. The antioxidant/ antiradical compounds possess the ability to provide electrons/ protons to the ABTS.+ radical. Thus, the discoloration of ABTS.+ occurs in the presence of antioxidant substances. The reduction of ABTS˙+ by the H-donating antioxidant substances was measured by the suppression of its characteristic absorption peak at 734 nm. The ABTS.+ assay offers extra flexibility that can be used at different pH levels; in contrast, the DPPH radical is sensitive to acidic pH. Further, ABTS˙+ is soluble in an aqueous and wide variety of organic solvents, hence useful for antioxidant assay of hydrophilic and hydrophobic candidates. Samples react rapidly with ABTS˙+ and attend a steady-state, whereas DPPH reacts very slowly and requires more incubation period. However, ABTS˙+ does not exist naturally; hence it should be generated chemically. The ABTS.+ scavenging activity of CDs was previously reported by various research groups [66,67,68,69]. The obtained IC50 value of PLACDs (0.051 mg/mL) for scavenging of ABTS.+ (Fig. 5e and Fig. 5f) is slightly low as compared to the PLLAE (0.059 mg/mL), which follows a similar trend as observed with scavenging of DPPH radical. The overall results of DPPH and ABTS.+ assay reflected the free radical scavenging potential of PLACDs.
The Ag3+, Hg2+, Cu2, As3+ cause slight quenching of the fluorescent intensity, whereas Fe3+ causes a severe quenching effect compared to control (without metal). The NH4+ ion causes a slight enhancement of fluorescence intensity, which may be due to the doping of metallic ions with the surface functional groups of PLACDs. The Mg2+, Ba2+, As5+, Ca2+, K+, and Na+ cause no change in intensity. The results display higher selectivity of PLACDs towards the sensing of Fe3+. The slight diminution of the fluorescence intensity in the presence of Ag3+, Hg2+, Cu2, and As3+ is attributed to the non-specific interaction of these metal ions with surface functional groups of PLACDs. In contrast, the higher selectivity towards Fe3+ is assigned to special coordination between –OH of PLACDs with Fe3+ [70]. The splitting of d orbital of Fe3+ occurs due to complexation with the phenolic –OH group of PLACDs and electrons from the excited state of PLACDs transferred to the d orbital of Fe3+, leading to quenching of fluorescent intensity [49, 71]. The Fe3+ sensing behavior of many CDs was reported previously [48, 49, 54, 70]. Yang's et al.synthesized highly fluorescent CDs by HTC method using citric acid and ethylenediamine as precursors. The authors have reported excellent selectivity and sensitivity of the CDs towards the Fe3+ ions with a limit of detection (LOD) of 18 μM [13]. In another work, Kailasa et al. reported the synthesis of CDs from tomato (Solanum lycopersicum) and investigated their sensitivity toward Fe3+ ions with a LOD of 0.016 μM [47]. Geetha et al. reported the synthesis of CDs from Boswellia ovalifoliolata bark extract by the HTC method and explored it as a potential Fe3+ ion sensing probe with a LOD value of 0.41 μM [48]. Edison et al.synthesized N-doped CDs from Prunus avium fruit extract by HTC method and reported its Fe3+ ion sensing property with LOD value of 0.96 μM [49]. Recently our research group also explored the Fe3+ ion sensing property of Tinospora cordifolia stem-derived carbon dots (TCSCDs) and Tinospora cordifolia leaves-derived carbon dots (TCLCDs) with LOD values of 0.725 μM and 0.414 μM μM, respectively [33]. Many of the researchers exploited the quenching phenomena of CDs upon the addition of Fe3+, and the quenching percentage could have corresponded to the concentrations of Fe3+. The quenching of the fluorescent intensity of PLACDs is possibly due to the interaction of its inherent surface functional groups and metal ions (Fe3+). The fluorescent quenching of CDs in the presence of metal ions (Fe3+) has been reported to follow various mechanisms, such as aggregation, electron or energy transfer, and dynamic quenching mechanism [71]. In the aggregation mechanism, the inherent massive amount of –OH groups on the surface of PLACDs makes a complex with metal ions and causes aggregation of PLACDs, which leads to quenching of fluorescent intensity [71]. In the electron transfer mechanism, the metal-fluorophore complexation occurs, followed by an electron transfer from the excited PLACDs to the metal ions. The quenching of fluorescence in such a mechanism is ascribed to the specific coordination interaction among the Fe3+ ions and the phenolic -OH groups of the PLACDs [71]. In the dynamic quenching mechanism, the excited PLACDs return back to the ground state due to bombardment between PLACDs and metal ions via charge transfer or energy transfer mechanism [72].
Nowadays, fluorescence-based sensors have received considerable attention due to their excellent sensitivity, quick response time, and low cost [13]. Theoretically, any alteration of fluorescence property, such as fluorescence intensity, emission wavelength, and anisotropy with respect to the concentration of various analytes, demonstrates the potential of CDs to be used as sensors [13]. The fluorescence changes of CDs used for sensing applications are governed by multiple mechanisms, such as photo-induced electron transfer (PET), resonance energy transfer (RET), photo-induced charge transfer (PCT), and inner filter effect (IFE). PET is a deactivation mechanism in which an internal redox reaction and complexation occurs among the excited state of the fluorophore (CDs) and analytes able to accept or donate an electron. Such complex mainly returns to the ground state without emission of a photon, thus decreasing the fluorescence intensity [13]. In RET, also known as Forster resonance energy transfer (FRET), a donor chromophore (CDs) which is in its excited state during returning to the ground state, simultaneously transfers energy to an acceptor chromophore (analyte) through nonradiative long-range dipolar interactions among excited donor and acceptor [13]. PCT involves the transfer of an electron among electron donor and acceptor functionalities to form a complex. The complexation leads to an alteration of electron energy levels and ultimately changes the fluorescence behavior [13]. IFE results from the absorption of excitation or emission light by absorbers in the detection system. Such phenomena occur when the absorption spectra of the absorbers overlap with the fluorescence excitation or emission spectra of fluorophores. Due to the unique possessions of CDs, they have been explored as sensors for a wide range of analytes, such as ions (Fe3+, Ag+, Hg2+, Cu2+, Cr6+, S2−, CN−), small molecules ( glutathione, cysteine, glucose, reactive oxygen and nitrogen species), macromolecules (proteins, DNA), and bacteria (Escherichia coli & Salmonella typhimurium) [13]. Among various metal cations, the Fe3+ ion is the most widely reported sensing target through fluorescent CDs in recent years [13]. Iron is an essential micronutrient for living systems, including human beings, plants, and animals [45, 46]. In the human body, the Fe3+ ion act as a key structural component or cofactor for various biochemical roles, such as electron transfer, oxygen transport, cell respiration, enzymatic reaction, DNA, and hemoglobin synthesis. The Fe3+ ion should be within the optimal physiologic range for proper biological functions. Its shortage and excess in a living body are the causative factors for various disorders, such as renal failure, anemia, heart disease, intelligence decline, Alzheimer’s disease, Parkinson’s, and cancer [47,48,49]. Therefore, it is very imperative to accurately and effectively analyze its level in biological and environmental samples to prevent harmful impacts on living beings. Currently used techniques (e.g., inductive coupled plasma-atomic emission spectroscopy, mass spectroscopy, amperometry, ion chromatography, plasmon resonance Raleigh scattering spectroscopy) for quantitative and qualitative analysis of Fe3+ ion require sophisticated instruments, expensive chemicals, long operational time, highly-trained personnel and tedious sample preparation procedures. Therefore, it is essential to develop cost-effective, easily available probes that can selectively sense Fe3+ ion accurately in the presence of other trace elements. Recently, CDs-based fluorescent probes have received substantial interest in identifying and quantifying many metal ions, including Fe3+ ion with a high degree of selectivity and sensitivity [40, 41, 47, 48].
The metal sensing results (Fig. 6) of PLACDs reflected high selectivity towards Fe3+ ion with its accurate estimation. Our synthesized PLACDs represents cost-effective, easily available probes that can selectively sense Fe3+ ion accurately in the presence of other trace elements. The LOD value (0.673 μM) for detection of Fe3+ ion is lower than and very close to the reported values of CDs from green sources, suggesting the higher sensitivity of PLACDs. Further, the LOD value is very lower than that of the maximum allowable limit for Fe3+ ion in drinking water (5.36 μM) as per the report of the World Health Organization (WHO), which can accurately estimate the quality of drinking water with respect to the Fe3+ ion content [48].
Conclusions
In the current study, we have synthesized green fluorescent carbon dots (PLACDs) from Piper longum leaves via a simple, uni-pot, and eco-friendly hydrothermal carbonization technique. The synthesized PLACDs are carbonaceous zero-dimensional quasi-spherical nanodots having excitation-dependent fluorescence property, pH-dependent fluorescence property, fluorescent ink property, excellent photostability, physical, chemical, and thermal stability. It has outstanding biocompatibility and concentration-dependent bioimaging activity in B16F10 (melanoma) and SiHa (cervical cancer) cell lines. The PLACDs exhibited excellent antiradical activity against DPPH and ABTS.+ compared to its precursor (PLLAE), reflecting its probable use as an antioxidant agent. Furthermore, they disclosed excellent sensitivity and selectivity to Fe3+ ion, which may be used as an effective probe for Fe3+ ion sensing. The excellent bacterial biocompatibility results of PLACDs may be useful for in vitro bacterial cell imaging. Merging its simple and eco-friendly uni-pot synthesis, excellent optical possessions, good biocompatibility, and long-term stability, the PLACDs holds great promise for its possible applications in cancer cell bioimaging, free radical scavenging, and Fe3+ ion sensing.
Data Availability
All data recorded and generated during this research are included in this article, and some of the data are also supplied as supplementary files.
Code Availability
All the data were produced using MS office package, a 21-day free trial version of Origin Pro 2021 (Microcal Software, Inc., Northampton, Northampton, MA, USA), and ImageJ software (National Institutes of Health, Bethesda, MD).
References
Z. Peng, X. Han, S. Li, A. O. Al-Youbi, A. S. Bashammakh, M. S. El-Shahawi, and R. M. Leblanc (2017). Coord. Chem. Rev. 343, 256.
L.-M. Shen and J. Liu (2016). Talanta 156, 245.
J. A. Jaleel and K. Pramod (2018). J. Control Release 269, 302.
M. A. Issa, Z. Z. Abidin, S. Sobri, S. A. Rashid, M. A. Mahdi, and N. A. Ibrahim (2020). J. Sci. Rep. 10 (1), 1.
P. Namdari, B. Negahdari, and A. Eatemadi (2017). Biomed. Pharmacother. 87, 209.
V. N. Mehta, S. Jha, and S. K. Kailasa (2014). Mater. Sci. Eng. C 38, 20.
J. R. Bhamore, S. Jha, T. J. Park, and S. K. Kailasa (2019). J. Photochem. Photobiol. B 191, 150.
T. Feng, X. Ai, G. An, P. Yang, and Y. Zhao (2016). ACS Nano 10 (5), 5587.
Q. Zeng, D. Shao, X. He, Z. Ren, W. Ji, C. Shan, S. Qu, J. Li, L. Chen, and Q. Li (2016). J. Mater. Chem. B 4 (30), 5119.
C. Liu, P. Zhang, X. Zhai, F. Tian, W. Li, J. Yang, Y. Liu, H. Wang, W. Wang, and W. Liu (2012). Biomaterials 33 (13), 3604.
S. L. D’souza, B. Deshmukh, K. A. Rawat, J. R. Bhamore, N. Lenka, and S. K. Kailasa (2016). New J. Chem. 40 (8), 7075.
D.-W. Zheng, B. Li, C.-X. Li, J.-X. Fan, Q. Lei, C. Li, Z. Xu, and X.-Z. Zhang (2016). ACS nano 10 (9), 8715.
X. Sun and Y. Lei (2017). Trends Analyt. Chem. 89, 163.
X. An, S. Zhuo, P. Zhang, and C. Zhu (2015). RSC Adv. 5 (26), 19853.
M.J. Meziani, X. Dong, L. Zhu, L.P. Jones, G.E. LeCroy, F. Yang, S. Wang, P. Wang, Y. Zhao, L. Yang, ACS Appl. Mater. Interfaces 8 (17), 10761 (2016)
R. Atchudan, T. N. J. I. Edison, K. R. Aseer, S. Perumal, and Y. R. Lee (2018). Colloids Surf. B Biointerfaces 169, 321.
I. Milenkovic, M. Algarra, C. Alcoholado, M. Cifuentes, J. M. Lázaro-Martínez, E. Rodríguez-Castellón, D. Mutavdžić, K. Radotić, and T. J. Bandosz (2019). Carbon 144, 791.
S. Zhao, M. Lan, X. Zhu, H. Xue, T.-W. Ng, X. Meng, C.-S. Lee, P. Wang, W. Zhang, ACS Appl. Mater. Interfaces 7(31), 17054 (2015)
P. Roy, P.-C. Chen, A. P. Periasamy, Y.-N. Chen, and H.-T. Chang (2015). Mater. Today 18 (8), 447.
Y. Han, D. Tang, Y. Yang, C. Li, W. Kong, H. Huang, Y. Liu, and Z. Kang (2015). Nanoscale 7 (14), 5955.
T. Feng, Q. Zeng, S. Lu, X. Yan, J. Liu, S. Tao, M. Yang, and B. Yang (2018). ACS Photonics 5 (2), 502.
A. N. Sahu and D. Mohapatra (2021). Nanomedicine 16 (28), 2491.
A.N. Sahu, D. Mohapatra, Herbal Drug Formulation and Standardization. (Ane Books Pvt. Ltd., 2021), p 1–10
D. Mohapatra, A. K. Agrawal, and A. N. Sahu (2021). J. Microencapsul. 38 (7–8), 594.
G. G. Naik, J. Shah, A. K. Balasubramaniam, and A. N. Sahu (2021). Nanomedicine 16 (7), 587.
S. A. A. Vandarkuzhali, V. Jeyalakshmi, G. Sivaraman, S. Singaravadivel, K. R. Krishnamurthy, and B. Viswanathan (2017). Sens. Actuators B Chem. 252, 894.
S. Manjusha, N. Parameswaran, and R. S. Malar (2018). Indo. Am. J. Pharm. Sci. 5 (4), 2942.
J. Das, D. Jha, R. Policegoudra, A. H. Mazumder, M. Das, P. Chattopadhyay, and L. Singh (2012). Indian J. Microbiol. 52 (4), 624.
T. Varughese, P.K. Unnikrishnan, M. Deepak, I. Balachandran, A. Rema Shree, J. Essent. Oil-Bear. Plants 19(1), 52 (2016)
J. Du, N. Xu, J. Fan, W. Sun, and X. Peng (2019). Small 15 (32), 1805087.
H. Li, X. Yan, D. Kong, R. Jin, C. Sun, D. Du, Y. Lin, and G. Lu (2020). Nanoscale Horiz. 5 (2), 218.
K. O. Boakye-Yiadom, S. Kesse, Y. Opoku-Damoah, M. S. Filli, M. Aquib, M. M. B. Joelle, M. A. Farooq, R. Mavlyanova, F. Raza, and R. Bavi (2019). Int. J. Pharm. 564, 308.
D. Mohapatra, M. B. Alam, V. Pandey, R. Pratap, P. K. Dubey, A. S. Parmar, and A. N. Sahu (2021). Nanomedicine 16 (23), 2039.
D. Mohapatra, R. Pratap, V. Pandey, P.K. Dubey, A.K. Agrawal, A.S. Parmar, A.N. Sahu, J. Fluoresc. 1 (2021)
K. Pavithra and S. J. F. S. Vadivukkarasi (2015). Food Sci. Hum. Wellness 4 (1), 42.
N. Maddu, In Antioxidants, (IntechOpen, 2019), p. 1–10
L. A. Pham-Huy, H. He, and C. Pham-Huy (2008). Int. J. Biomed. Sci. 4 (2), 89.
G. G. Naik, M. B. Alam, V. Pandey, D. Mohapatra, P. K. Dubey, A. S. Parmar, and A. N. Sahu (2020). J. Fluoresc. 30 (2), 407.
K. K. Gudimella, T. Appidi, H.-F. Wu, V. Battula, A. Jogdand, A. K. Rengan, and G. Gedda (2021). Colloids Surf. B 197.
J. Jia, B. Lin, Y. Gao, Y. Jiao, L. Li, C. Dong, and S. Shuang (2019). Spectrochim. Acta A 211, 363.
T. Pal, S. Mohiyuddin, and G. Packirisamy (2018). ACS omega 3 (1), 831.
D. Yang, L. Li, L. Cao, Y. Zhang, M. Ge, R. Yan, W.-F. Dong, Analyst 146 (7) (2021)
X. Zhang, H. Wang, C. Ma, N. Niu, Z. Chen, S. Liu, J. Li, and S. Li (2018). Mater. Chem. Front. 2 (7), 1269.
A. Bayat, S. Masoum, and E. S. Hosseini (2019). J. Mol. Liq. 281, 134.
X. Ding, L. Song, Y. Han, Y. Wang, X. Tang, G. Cui, Z. Xu, Biomed. Res. Int. 2019, (2019)
G. R. Rout and S. Sahoo (2015). Rev. Agric. Sci. 3, 1.
S. K. Kailasa, S. Ha, S. H. Baek, S. Kim, K. Kwak, and T. J. Park (2019). Mater. Sci. Eng. C 98, 834.
G. Venkatesan, V. Rajagopalan, and S. N. Chakravarthula (2019). J. Environ. Chem. Eng. 7 (2).
T. N. J. I. Edison, R. Atchudan, J.-J. Shim, S. Kalimuthu, B.-C. Ahn, and Y. R. Lee (2016). J. Photochem. Photobiol. B 158, 235.
U. H. A. M. Hazli, A. Abdul-Aziz, S. Mat-Junit, C. F. Chee, and K. W. Kong (2019). Food Res. Int. 115, 241.
E.A. Shalaby, S.M. Shanab, Indian J. Geomarine Sci. (2013)
A.A. Shahat, S. Hidayathulla, A.A. Khan, A.M. Alanazi, O.T. Al Meanazel, A.S. Alqahtani, M.S. Alsaid, A.A. Hussein, Acta. Trop. 191, 243 (2019)
M. A. Jhonsi, D. A. Ananth, G. Nambirajan, T. Sivasudha, R. Yamini, S. Bera, and A. Kathiravan (2018). Spectrochim. Acta A 196, 295.
A. Sachdev and P. Gopinath (2015). Analyst 140 (12), 4260.
C. J. Jeong, A. K. Roy, S. H. Kim, J.-E. Lee, J. H. Jeong, I. In, and S. Y. Park (2014). Nanoscale 6 (24), 15196.
M. Thakur, S. Pandey, A. Mewada, V. Patil, M. Khade, E. Goshi, M. Sharon, J. Drug Deliv. (2014)
W. Li, Z. Yue, C. Wang, W. Zhang, and G. Liu (2013). RSC Adv. 3 (43), 20662.
V. N. Mehta, S. Jha, R. K. Singhal, and S. K. Kailasa (2014). New J. Chem. 38 (12), 6152.
K. Hola, A. B. Bourlinos, O. Kozak, K. Berka, K. M. Siskova, M. Havrdova, J. Tucek, K. Safarova, M. Otyepka, and E. P. Giannelis (2014). Carbon 70, 279.
V. Arul, T. N. J. I. Edison, Y. R. Lee, and M. G. Sethuraman (2017). J. Photochem. Photobiol. B 168, 142.
P. C. Senapati, S. K. Sahoo, and A. N. Sahu (2016). Biomed. Pharmacother. 80, 42.
L. Wu, J. Zhang, and W. Watanabe (2011). Adv. Drug Deliv. Rev. 63 (6), 456.
A. Mewada, S. Pandey, S. Shinde, N. Mishra, G. Oza, M. Thakur, M. Sharon, and M. Sharon (2013). Mater. Sci. Eng. C 33 (5), 2914.
W. Zhang, Z. Zeng, and J. Wei (2017). J. Phys. Chem. C 121 (34), 18635.
N. Sharma, G. S. Das, and K. J. A. M. Yun (2020). Biotechnology 104 (16), 7187.
S. Roy, P. Ezati, J.-W. Rhim, and A. C. S. Appl (2021). Polym. Mater. 3 (12), 6437.
C. Dong, S. Wang, M. Ma, P. Wei, Y. Chen, A. Wu, Z. Zha, and H. Bi (2021). Appl. Mater. Today 25.
J. Zhao, H. Wang, H. Geng, Q. Yang, Y. Tong, W. He, and A. C. S. Appl (2021). Nano Mater. 4 (7), 7253.
Z. T. Rosenkrans, T. Sun, D. Jiang, W. Chen, T. E. Barnhart, Z. Zhang, C. A. Ferreira, X. Wang, J. W. Engle, and P. Huang (2020). Adv. Sci. 7 (12), 2000420.
X. Gong, W. Lu, M. C. Paau, Q. Hu, X. Wu, S. Shuang, C. Dong, and M. M. Choi (2015). Anal. Chim. Acta 861, 74.
Y. Dong, J. Cai, X. You, and Y. Chi (2015). Analyst 140 (22), 7468.
M. J. Molaei (2020). Anal. Methods 12 (10), 1266.
Acknowledgements
The authors are thankful to the Department of Pharmaceutical Engineering & Technology, IIT (BHU); Department of Physics, IIT (BHU); Centre for Genetics Disorders, Institute of Science, Banaras Hindu University, and Central Instrument Facility, IIT (BHU), Varanasi, India for providing infrastructural and instrumental services. The microbiological facilities provided by the Department of Microbiology, IMS (BHU), Varanasi, India, are also greatly acknowledged.
Funding
The financial support for this work was provided as a scholarship to Debadatta Mohapatra by the Ministry of Human Resource Development (MHRD), Government of India. Author Alakh N. Sahu is thankful to the Department of Biotechnology (DBT), Ministry of Science & Technology, Government of India, New Delhi, India, for providing the funding (Sanction order No. BT/PR25498/NER/95/1223/2017) for exploring phytochemical and pharmacological evaluations of bioactivity guided fractions of medicinal plants of Tripura.
Author information
Authors and Affiliations
Contributions
Debadatta Mohapatra executed most of the experimental works, data collection, processing, analysis, validation, interpretation, and wrote the original manuscript. Ravi Pratap, Vivek Pandey, and Singh Shreya contributed to the experimentation, reviewed, edited, and scientifically modified the manuscript. Alakh N. Sahu, Avanish S. Parmar, Pawan K. Dubey, and Prakash Ch. Senapati contributed to supervision, interpretation of data & concluding the results, technical support, manuscript review, and editing functions.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical Approval
Approval not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Mohapatra, D., Pratap, R., Pandey, V. et al. In Vitro Cancer Cell Imaging, Free Radical Scavenging, and Fe3+ Sensing Activity of Green Synthesized Carbon Dots from Leaves of Piper longum. J Clust Sci 34, 1269–1290 (2023). https://doi.org/10.1007/s10876-022-02303-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10876-022-02303-9