
Abstract
A new Zr-substituted polyoxotungstate, [H2N(CH3)2]11KNa5H7[Zr4K2(β-GeW10O38)2(A-α-PW9O34)2]·39H2O (1), has been hydrothermally synthesized and characterized by single crystal X-ray diffraction, IR spectrum, UV–vis diffuse-reflectance spectrum, elemental analysis, powder X-ray diffraction, ICP-MS test and thermogravimetric analysis. The prominent feature of 1 is that mixed Keggin-type dilacunary [β-GeW10O38]12− fragment and trilacunary [A-α-PW9O34]9− fragment construct the basic framework of 1. And 1 displays special 1D double-chain and 2D thick layer formed by K+/Na+ ions. 1 exhibits well electrocatalytic behavior in the reduction of BrO3−. Also, the experimental results of catalytic oxidation of representative thioethers manifest that 1 possesses a good catalytic capacity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Polyoxometalates (POMs) are followed closely by virtue of diverse structures and electronic properties, leading to flexible and multiple applications in energy conversion, catalysis, biomedical research as well as design and self-assembly of new materials [1,2,3,4,5,6,7,8,9,10,11,12,13]. As a significant branch of POMs, transition-metal-substituted polyoxometalates (TMSPs) own characteristic physicochemical properties, like thermodynamic stability to oxidation and tunability of redox properties [14]. Among them, ZrIV-substituted POMs (ZrSPs) have been developing uninterruptedly since the first Zr-substituted POM [Zr3(μ-OH)3(A-β-SiW9O34)2]11− was reported in 1989 [15]. It´s worth to note that ZrIV ions have a higher oxidation state (+ 4), variable coordination modes (6, 7 and 8-coordinated modes) and strong oxytropic property [16], resulting in distinctive structures in the reaction of ZrIV ions and POMs. Of the ZrSPs that have been made, the most classic sandwich configuration has become a common occurrence, such as [Zr3O(OH)2(α-GeW9O34)(β-GeW9O34)]12− [17], [Zr4O2(OH)2(H2O)4(β-SiW10O37)2]10− [18] and [Zr3(μ2-OH)2(μ2-O)(A-α-GeW9O34)(1,4,9-α-P2W15O56)]14− [19]. Some ZrSPs functionalized by organic ligands were also reported, for instance, the acetate-containing [Zr6O4(OH)4(H2O)2(CH3COO)5(AsW9O33)2]11− [20] and the chiral tartrate functionalized ZrSPs {[α-P2W15O55(H2O)]Zr3(μ-O)(H2O)(L/D-tartH)[α-P2W16O59]}15− [21]. Apart from these, some high-nuclear and multimeric ZrSPs were also made, for example, the ribbon trimer [Zr6O2(OH)4(H2O)3(β-SiW10O37)3]14− [18], the staggered tetramer [{Zr5(µ3-OH)4(OH)2}@{Zr2(OAc)2(α-GeW10O38)2}2]24− [22] and the giant Zr24-containing ZrSP [Zr24O22(OH)10(H2O)2(W2O10H)2(GeW9O34)4(GeW8O31)2]32− [23]. Notice that the maximum number of Zr atoms in ZrSPs made by the aqueous solution method is only 6 [18, 20, 24], while the numbers of Zr atoms in ZrSPs is up to 8 [25], 9 [22] and 24 [23] under hydrothermal conditions. In fact, the hydrothermal technique has been proved to be a unique way of making ZrSPs for incomparable conditions, like capturing intermediate phases easily, increasing the solubilities of materials and conducive to the formation of high-quality and stable crystals [26,27,28,29]. So far, a series of novel TMSPs have been made in our group under hydrothermal conditions [26, 30,31,32,33,34]. As our important research subject, some novel ZrSPs have been made under hydrothermal conditions [19, 22, 23, 25, 32,33,34].
Here, a new ZrSP built by two types of lacunary fragments, [H2N(CH3)2]11KNa5H7[Zr4K2(β-GeW10O38)2(A-α-PW9O34)2]·39H2O (1), has been made under hydrothermal conditions, showing a 1D double-chain and further expands into 2D thick layer. Electrocatalytic reduction of BrO3− and catalytic oxidation of sulfide to sulfoxide/sulfoxide were also carried out, both of which show excellent catalytic activity.
Experimental Section
Materials and Methods
Na9[A-α-PW9O34]·7H2O [35] and K8Na2[A-α-GeW9O34]·25H2O [36] were prepared according to the literature. All other reagents were purchased commercially and used without further purification. IR spectrum was recorded on a Smart Omni-Transmission spectrometer from 400 to 4000 cm−1. UV–vis diffuse-reflectance spectrum was measured by using a Perkin Elmer Lambda 900 UV/vis spectrophoto- meter at room temperature. Elemental analyses were performed on a EuroEA3000 elemental analyzer. Power X-ray diffraction patterns were recorded using a Bruker D8 Advance X-ray diffractometer with Cu Kα radiation (λ = 1.54056 Å). Thermogravimetric analysis (TGA) was measured on a Mettler Toledo TGA/DSC 1000 analyzer in an air atmosphere with a heating rate of 10 °C min−1. The DH 7000 electrochemical workstation was used for electrochemical experiments. Inductively coupled plasma mass spectrometry was measured on a Thermo Scientific iCAP Q ICP-MS spectrometer.
Synthesis of 1
Na9[A-α-PW9O34]·7H2O (1.000 g, 0.390 mmol), K8Na2[A-α-GeW9O34]·25H2O (0.500 g, 0.162 mmol), ZrOCl2·8H2O (0.205 g, 0.636 mmol), NaCl (0.500 g, 8.555 mmol) and dimethylamine hydrochloride (0.400 g, 4.906 mmol) were stirring in 10 mL H2O for 1 h. The pH of the mixture was adjusted to 5.5 with 1 mol L−1 HCl. Stirring for another 5 min, then the resulting mixture was sealed in a 25 mL Teflon-lined steel autoclave and heated at 140 °C for 5 days. The colorless block crystals of 1 were obtained after cooling to room temperature and filtering. Yield: 15.70% (based on Zr). Elemental analyses calcd for 1 (%): C, 2.34; H, 1.53; N, 1.36; Ge, 1.28; P, 0.55; W, 61.76; Zr, 3.23; K, 1.03; Na, 1.02. Found: C, 2.74; H, 1.51; N, 1.39; Ge, 1.35; P, 0.53; W, 59.92; Zr, 3.28; K, 2.51; Na, 1.78.
Experimental Procedure of Oxidation Various Aromatic Thioethers Catalyzed by 1
The catalytic oxidation of various aromatic thioethers was carried out in a 25 mL glass vessel under magnetic stirring at 60 °C. A certain amount of catalyst 1, a solution containing thioether, acetonitrile, 30% H2O2 and dodecane (an internal standard for GC-analysis) were put into the glass vessel, then the glass vessel was placed into a thermostatted oil-bath (60 °C) for reaction. The vessel was cooled to room temperature after reaction, supernatant was extracted with dichloromethane for GC-analysis. The thioether oxidation products (sulfoxide and sulfone) were identified with GC and quantified using gas chromatography with internal standard techniques.
Cyclic Experiment of Catalyst 1
The oxidation of methyl phenyl sulfide (entry 1 in Table 2) was selected as a probe reaction. At first, 0.5 mmol of methyl phenyl sulfide, 1.5 mmol of H2O2, and 0.5 × 10–2 mmol of 1 were added into 5 mL of MeCN, then the mixture was stirred at 60 °C for 1 h. Upon the completion of the reaction, the catalyst was collected by filtration and dried. This process was carried out repeatedly.
X-Ray Crystallographic Analysis
Single crystal X-ray diffraction data of 1 was collected on a Gemini A Ultra diffractometer with graphite monochromated MoKα (λ = 0.71073 Å) at room temperature. The program SADABS was used for the absorption correction. The direct method was adopted to determine the structure and full-matrix least-squares method on F2 to refine with SHELXTL-2014 program package. No hydrogen atoms associated with water molecules were located from the difference Fourier map. These highly disordered solvent molecules in crystal structures are difficult to be determined by X-ray diffraction, resulting in the presence of the solvent accessible voids which were treated using the SQUEEZE method in PLATON. Therefore, another 30 lattice water molecules and 7 [H2N(CH3)2]+ are estimated from elemental analysis and TGA. On the basis of charge-balance considerations, 7 protons are directly added to the molecular formula. These phenomena are common in POM chemistry [37,38,39,40]. Crystallographic data and structure refinement parameters for 1 are listed in Table 1. CCDC 2050011 contains the supplementary crystallographic data for 1 and this data is available free of charge at The Cambridge Crystallographic Data Centre.
Results and Discussion
Crystal Structure of 1
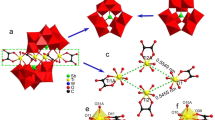
1 crystallizes in the triclinic space group P-1 and contains a polyoxoanion [Zr4K2(β-GeW10O38)2(A-α-PW9O34)2]24– (1a, Fig. 1f/2a), 5 Na+ ions, 1 K+ ion, 11 monoprotonated [H2N(CH3)2]+ cations, 9 coordinated water molecules and 30 lattice water molecules. Bond valence sum (BVS) calculations [41] indicate that the oxidation state of all Zr, Ge and P atoms are + 4, + 4 and + 5, respectively. Two kinds of trilacunary precursors were added in the reaction, of which only the transformation of [A-α-GeW9O34]10− → [β-GeW10O38]12− (Fig. 1a/d) happened while [A-α-PW9O34]9− (Fig. 1b) keep the configuration. We speculate that there may be a decomposition and recombination of [A-α-GeW9O34]10− unit. The 1a (Fig. 1f) is built from two kinds of fragments, [Zr2(β-GeW10O38)]2− (Fig. 1e) and [K(A-α-PW9O34)]8− (Fig. 1c), which are connected alternately by µ3-O/µ-O. Two Zr4+ ions locate at two vacant sites of [β-GeW10O38]12− unit to form the [Zr2(β-GeW10O38)]2− moiety and one K+ ion occupies two vacant sites of [A-α-PW9O34]9− unit to form the [K(A-α-PW9O34)]8− moiety. To our knowledge, the coexistence of two different types of lacunary fragments in one molecular structure is quite rare.

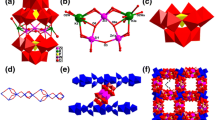
(a) Polyhedral view of A-α-XW9O34 (X = P/Ge). (b–e) Polyhedral/ball-and-stick view of [A-α-PW9O34]9−, [K(A-α-PW9O34)]8−, [β-GeW10O38]12− and [Zr2(β-GeW10O38)]2−. (f) Polyhedral/ball-and-stick representation of polyoxoanion 1a. Color code: Zr, green; K, turquoise; Ge, yellow; O, red; H2O, rose; WO6, red; GeO4, yellow; PO4, purple (Color figure online)
As shown in Fig. 2, it can be found that a ring-shaped octa-nuclear cluster {Zr4K2W2} (Fig. 2b) emerges in the structure. Four Zr4+, two W and two K+ atoms connect alternately by bridging oxygen constituting the octa-nuclear {Zr4K2W2} cluster. Four Zr4+ ions are isolated without Zr–O–Zr linkages. The introduction of two K+ ions makes the structure 1a more stable. All the four independent Zr4+ ions adopt a heptacoordinate mode, exhibiting the distorted capped octahedral geometry, each of which is attached to five O atoms from [β-GeW10O38]12− unit and two O atoms from [A-α-PW9O34]9− unit. Moreover, all the Zr–O bond lengths are in the range of 2.066(15)–2.463(14) Å. K1+ and K2+ ions are octacoordinated by three O atoms from [A-α-PW9O34]9− unit, three O atoms from two [β-GeW10O38]12− units and two coordinated water molecules. K–O bond lengths vary from 2.742(14) to 3.213(15) Å.

(a) Ball-and-stick view of 1a. (b) Ball-and-stick view of the ring-shaped octa-nuclear {Zr4K2Ge2} cluster. Color code: Zr, green; K, turquoise; O, red; H2O, rose; W, black (Color figure online)
The alkali metal K3+ ions link three neighboring 1a to generate a 1D double-chain along the a-axis (Fig. 3a). Furthermore, every adjacent 1D double-chain is interconnected by Na4+ ions along the b-axis, forming a 2D thick layer (Fig. 3b, S1). The simplified representations of 1D double-chain and 2D thick layer are shown in Fig. 4a, b, respectively.

(a) The 1D double-chain connected by K3+ ions; (b) the 2D thick layer constructed from 1D double-chain linked by Na4+ ions. Color code: Na, bule; WO6, red; GeO4, yellow; PO4, purple; ZrO7, green; KO8, turquoise. Part of the Na atoms, and lattice water molecules are omitted for clarity (Color figure online)

(a) Simplified representation of 1D double-chain. (b) Simplified representation of 2D thick layer. Color code: red balls, polyoxoanion 1a; turquoise balls, K3+; blue balls, Na4+; purple rod, K–O bond; green rod, Na–O bond (Color figure online)
Electrochemical Properties
POMs are famous for their excellent redox ability and structural stability, behaving as the participation in multi-electron transfer with structures unchanged [5, 42]. For the past few years, the application of TMSPs in electrochemical and electrocatalytic properties have been reported in an endless stream [43,44,45]. Here we also did the corresponding research. The electrochemical performances of 1-based carbon paste electrode (1-CPE) were carried out in a trielectrode system with 0.5 M Na2SO4/H2SO4 (pH 5.70) aqueous solution. The cyclic voltammetry (CV) curves of 1-CPE in the potential window range of – 1500 to 500 mV (potentials vs Ag/AgCl) at scanning rates from 50 to 500 mV s−1 are shown in Fig. 5b. At the rate of 100 mV s−1 (Fig. 5a), 1-CPE shows two pairs of redox peaks − 437 mV and − 57 mV (I, I′), − 1141 mV and − 943 mV (II, II′) in the potential range from − 1500 to 500 mV, the mean peak potentials E1/2 = (Epa + Epc)/2 are − 247 mV (I–I′) and − 1042 mV (II–II′) correspondingly (vs. the Ag/AgCl electrode). All the two peaks are assigned to the redox of WVI centers, the redox peaks of the Zr4+ ions are silent under this testing conditions. With the scan rate increases gradually, it’s clearly observed that the cathodic peak potentials move to the negative direction and the anodic peak potentials move to the positive direction (Fig. 5b). The peak current has a linear correlation with the scanning rate, manifesting the redox processes of 1-CPE are surface-controlled.

(a) CV of 1-CPE in 0.5 M Na2SO4 + H2SO4 solution (pH 5.70) at 100 mV s−1; (b) CVs of 1-CPE at different scan rates (from inner to outer: 50, 100, 150, 200, 250, 300, 350, 400, 450 and 500 mV s−1; inset is the variation of the anodic peak current II′ with the scan rate); (c) the evolution of CV of 1-CPE in 0.5 M Na2SO4 + H2SO4 solution (pH 5.70) containing various BrO3− concentrations (0, 0.4, 0.8, 1.2, 1.6 mmol/L), scan speed: 100 mV s−1; (d) CAT versus concentrations of BrO3−
Moreover, the electrocatalytic behavior of 1-CPE in the reduction of BrO3− was studied in 0.5 M Na2SO4 + H2SO4 solution (pH 5.70) at a scan speed of 100 mV s−1. With the increase of concentrations of BrO3−, the reduction peak currents show a trend of increase while the oxidation peak currents show a downward trend (Fig. 5c). On the basis of the reduction peak current II, the catalytic efficiencies (CATs) [46] of 1-CPE at BrO3− concentrations of 0, 0.4, 0.8, 1.2, 1.6 mmol/L are calculated as 0, 19.65%, 44.12%, 72.77% and 108.78% (Fig. 5d). Hence, 1-CPE has effective electrocatalytic activity on the reduction of BrO3−.
Catalytic Oxidation of Thioethers
Sulfone and sulfoxide compounds have extensive application prospects due to their biological activities [47], simultaneously, they are important intermediates in basic organic synthesis and other industries [48,49,50]. Direct oxidation of thioethers is one of the main methods to prepare sulfone and sulfoxide. H2O2 has attracted much attention as a cleaning oxidant in oxidation of thioethers [51], meanwhile, POMs catalysts have been investigated a lot as catalyst of thioethers oxidation reactions [20, 52, 53]. Here, 1 was used as a heterogeneous catalyst in the reaction of oxidation various aromatic thioethers with 30% H2O2 (Scheme 1). The oxidation products of thioether were identified by GC. Both of the selectivity of sulfone and sulfoxide and the conversion rate of initial sulfide were tested by GC. Referring to the catalytic conditions of Zr24-containing POM as the catalyst to catalyze organic thioethers reported by our group [23], we carried out the tests under the following conditions: 0.5 mmol of substrate, 1.5 mmol of H2O2, 0.5 × 10–2 mmol of 1, 5 mL of acetonitrile, 60 °C and 1 h reaction time.

Catalytic oxidation of thioethers
The oxidation reaction data of six representative aromatic thioethers in MeCN at 60 °C in the presence of 1 are gathered in Table 2. Thereinto, entries 1–5 manifest the complete transformation of substrates and the selectivity of 100% sulfone. Dibenzothiophene (entry 6) with large steric hindrance has a slightly lower conversion rate of 99%, but still show the selectivity of 100% sulfone. Hence, 1 is effective catalyst in the process of oxidation thioethers with H2O2. 1 has better catalytic effect under the same conditions compared to the work reported by our group [23]. The cyclic experiment was carried out with methyl phenyl sulfide (entry 1) as the model substrate under the same condition. At the end of each cyclic experiment, the catalyst was collected by filtration and reclaimed for the next experiment. It can be seen from the data of cyclic experiment (Table S1) that the catalytic activity of 1 changed little after five cycles. Comparison of IR spectrum of the pristine catalyst 1 and the recovered catalyst after the fifth cyclic experiment (Fig. S2), the basic skeleton structure is well maintained. Some of the slight differences in the comparison of IR spectrum may be due to the oxidation of recovered samples by H2O2 [33].
IR Spectrum
IR spectrum of 1 (Fig. S3) was obtained within the scope of 4000–400 cm−1 and shows the characteristic vibration patterns from polyoxotungstate clusters, dimethylamine and water molecules. In the low wavenumber region, the absorption peaks at 871 cm−1 and 1074 cm−1 can be attributed to υ(Ge–Oa) and υ(P–Oa) stretching vibration respectively. The other three characteristic absorption peaks at 943, 797, and 705 cm−1 are assigned to υ(W–Ot), υ(W–Ob) and υ(W–Oc) stretching vibrations, severally [19]. In the high wavenumber region, the absorption peaks at 3444 and 1628 cm−1 are corresponding to the stretching and bending vibrations of O–H in the water molecules. Besides, the N–H, C–H and C–N stretching vibrations absorption peaks from dimethylamine molecules are found at 3142, 2790, and 1466 cm−1 [32, 53]. So, the results of IR spectrum of 1 agree well with X-ray diffraction structural analyses.
UV–Vis Analysis
The solid-state UV–Vis diffuse-reflectance spectrum of 1 has been texted (Fig. S4). Based on the Kubelka–Munk equation α/S = (1–R)2/2R, where α is the K–M absorption coefficient, S is the K–M scattering coefficient and R is the relative reflectance to BaSO4. The band gap (Eg) of 1 is estimated to be 3.51 eV, which can be determined by extrapolation from the linear portion of the absorption edge in a α/S versus energy plot. The band gap value manifests that 1 belongs to a wide-gap semiconductor.
Powder X-Ray Diffraction
The experimental PXRD patterns of the samples 1 are consistent well with this simulated by the single crystal X-ray diffraction, proving that the phase purity of the samples is good. Due to the change of preferred orientation of powder samples during experiment, the experimental and simulated PXRD patterns show differences of intensity (Fig. S5).
Conclusions
In summary, a new ZrSP [H2N(CH3)2]11KNa5H7[Zr4K2(β-GeW10O38)2(A-α-PW9O34)2]·39H2O built by two different lacunary fragments of [A-α-PW9O34]9− and [β-GeW10O38]12− was successfully made under hydrothermal conditions. It shows good electrocatalytic effect on the reduction of BrO3− and high conversion rate and selectivity of 100% sulfone in the catalytic oxidation of aromatic thioethers with H2O2 as well as superior recyclability. Further works on making novel ZrSPs are ongoing in our group.
Supporting Information
X-ray crystallographic file (CIF); selected bond distances, IR spectrum, TG curve, Powder X-ray diffraction and UV–Vis diffuse-reflectance spectrum for the title compound.
References
A. Misra, K. Kozma, C. Streb, and M. Nyman (2019). Angew. Chem. Int. Ed. 58, 2.
S. T. Zheng and G. Y. Yang (2012). Chem. Soc. Rev. 41, 7623.
S. S. Wang and G. Y. Yang (2015). Chem. Rev. 115, 4893.
W. H. Fang and G. Y. Yang (2018). Acc. Chem. Res. 51, 2888.
N. Li, J. Liu, B. X. Dong, and Y. Q. Lan (2020). Angew. Chem. Int. Ed. 59, 20779.
J. X. Liu, X. B. Zhang, Y. L. Li, S. L. Huang, and G. Y. Yang (2020). Coord. Chem. Rev. 414, 213260.
I. A. Weinstock, R. E. Schreiber, and R. Neumann (2018). Chem. Rev. 118, 2680.
J. S. Lee, C. Lee, J. Y. Lee, J. Ryu, and W. H. Ryu (2018). ACS Catal. 8, 7213.
Z. Zhou, D. D. Zhang, L. Yang, P. T. Ma, Y. A. Si, U. Kortz, J. Y. Niu, and J. P. Wang (2013). Chem. Commun. 49, 5189.
K. Yang, Y. X. Ying, L. L. Cui, J. C. Sun, H. Luo, Y. Y. Hu, and J. W. Zhao (2021). Energy Storage Mater. 34, 203.
D. Wang, L. L. Liu, J. Jiang, L. J. Chen, and J. W. Zhao (2020). Nanoscale 12, 5705.
X. Xu, R. R. Meng, C. T. Lu, L. Mei, L. J. Chen, and J. W. Zhao (2020). Inorg. Chem. 59, 3954.
B. X. Zeng, Y. Zhang, Y. H. Chen, G. P. Liu, Y. Z. Li, L. J. Chen, and J. W. Zhao (2021). Inorg. Chem. 60, 2663.
O. A. Kholdeeva, G. M. Maksimov, R. I. Maksimovskaya, M. P. Vanina, T. A. Trubitsina, D. Y. Naumov, B. A. Kolesov, N. S. Antonova, J. J. CarbÓ, and J. M. Poblet (2006). Inorg. Chem. 45, 7224.
R. G. Finke, B. Rapko, and T. J. R. Weakly (1989). Inorg. Chem. 28, 1573.
D. Li, H. Han, Y. Wang, X. Wang, Y. Li, and E. Wang (2013). Eur. J. Inorg. Chem. 10–11, 1926.
L. L. Chen, L. L. Li, B. Liu, G. L. Xue, H. M. Hua, F. Fu, and J. W. Wang (2009). Inorg. Chem. Commun. 12, 1035.
B. S. Bassil, M. H. Dickman, and U. Kortz (2006). Inorg. Chem. 45, 2394.
Z. Zhang, J. W. Zhao, and G. Y. Yang (2017). Eur. J. Inorg. Chem. 26, 3244.
G. Al-Kadamany, S. S. Mal, B. Milev, B. G. Donoeva, R. I. Maksimovskaya, O. A. Kholdeeva, and U. Kortz (2010). Chem. Eur. J. 16, 11797.
X. Fang, T. M. Anderson, and C. L. Hill (2005). Angew. Chem. Int. Ed. 44, 3540.
Z. Zhang, H. L. Li, Y. L. Wang, and G. Y. Yang (2020). Inorg. Chem. 58, 2372.
L. Huang, S. S. Wang, J. W. Zhao, L. Cheng, and G. Y. Yang (2014). J. Am. Chem. Soc. 136, 7637.
B. S. Bassil, S. S. Mal, M. H. Dickman, U. Kortz, H. Oelrich, and L. Walder (2008). J. Am. Chem. Soc. 130, 6696.
Z. Zhang, Y. L. Wang, Y. Liu, S. L. Huang, and G. Y. Yang (2020). Nanoscale. 12, 18333.
J. W. Zhao, H. P. Jia, J. Zhang, S. T. Zheng, and G. Y. Yang (2007). Chem. Eur. J. 13, 10030.
J. Gopalakrishnan (1995). Chem. Mater. 7, 1265.
B. Nohra, H. E. Moll, L. M. R. Albelo, P. Mialane, J. Marrot, C. M. Draznieks, M. O’Keeffe, R. N. Biboum, J. Lemaire, B. Keita, L. Nadjo, and A. Dolbecq (2011). J. Am. Chem. Soc. 133, 13363.
H. N. Miras, J. Yan, D. L. Long, and L. Cronin (2012). Chem. Soc. Rev. 41, 7403.
S. T. Zheng, J. Zhang, X. X. Li, W. H. Fang, and G. Y. Yang (2010). J. Am. Chem. Soc. 132, 15102.
S. T. Zheng, J. Zhang, and G. Y. Yang (2008). Angew. Chem. Int. Ed. 47, 3909.
H. L. Li, C. Lian, D. P. Yin, and G.-Y. Yang (2020). Inorg. Chem. 59, 12842.
Y. L. Wang, Z. Zhang, H. L. Li, X. Y. Li, and G. Y. Yang (2019). Eur. J. Inorg. Chem. 3–4, 417.
Y. L. Wang, J. W. Zhao, Z. Zhang, J. J. Sun, X. Y. Li, B. F. Yang, and G. Y. Yang (2019). Inorg. Chem. 58, 4657.
A. P. Ginsberg (1990). Inorg. Synth. 27, 108.
L. H. Bi, U. Kortz, S. Nellutla, A. C. Stowe, J. Tol, N. S. Dalal, B. Keita, and L. Nadjo (2005). Inorg. Chem. 44, 896.
F. Bannani, S. Floquet, N. Leclerc-Laronze, M. Haouas, F. Taulelle, J. Marrot, P. Koerler, and E. Cadot (2012). J. Am. Chem. Soc. 134, 19342.
R. S. Winter, J. M. Cameron, and L. Cronin (2014). J. Am. Chem. Soc. 136, 12753.
H. Xue, Z. Zhang, R. Pan, B. F. Yang, H. S. Liu, and G. Y. Yang (2016). CrystEngComm. 18, 4643.
G. J. Cao, J. D. Liu, T. T. Zhuang, X. H. Cai, and S. T. Zheng (2015). Chem. Commun. 51, 2048.
B. I. D. Brown and D. Altermatt (1985). Acta. Crystallogr. B41, 244.
X. Y. Ma, K. Yu, J. Yuan, L. P. Cui, J. H. Lv, W. T. Dai, and B. B. Zhou (2020). Inorg. Chem. 59, 5149.
M. Ibrahim, Y. X. Xiang, B. S. Bassil, Y. H. Lan, A. K. Powell, P. D. Oliveira, B. Keita, and U. Kortz (2013). Inorg. Chem. 52, 8399.
A. Haider, B. S. Bassil, J. S. López, H. M. Qasim, C. S. D. Pipaón, M. Ibrahim, D. Dutta, Y. S. Koo, J. J. Carbó, J. M. Poblet, J. G. Mascarós, and U. Kortz (2019). Inorg. Chem. 58, 11308.
S. J. Folkman, J. S. Lopez, J. G. Mascarós, and R. G. Finke (2018). J. Am. Chem. Soc. 140, 12040.
B. Keita, A. Belhouaria, L. Nadjo, and R. J. Contant (1995). Electroanal. Chem. 381, 243.
M. C. Carreno (1995). Chem. Rev. 95, 1717.
I. Fernández and N. Khiar (2003). Chem. Rev. 103, 3651.
V. Y. Kukushkin (1995). Coord. Chem. Rev. 139, 375.
J. Song, Z. Luo, D. K. Britt, H. Furukawa, O. M. Yaghi, K. I. Hardcastle, and C. L. Hill (2011). J. Am. Chem. Soc. 133, 16839.
M. R. Mauryaa, A. K. Chandrakar, and S. Chandb (2007). J. Mol. Catal. A: Chem. 274, 192.
M. Y. Yu, J. Yang, T. T. Guo, and J. F. Ma (2020). Inorg. Chem. 59, 4985.
H. L. Li, C. Lian, L. J. Chen, J. W. Zhao, and G. Y. Yang (2020). Nanoscale. 12, 16091.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 21831001, 21571016, 91122028) and the National Natural Science Foundation of China for Distinguished Young Scholars (No. 20725101).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Sheng, YH., Sun, JJ., Zhang, PY. et al. A Zr-Substituted Polyoxotungstate Built by [β-GeW10O38]12− and [A-α-PW9O34]9− Fragments: Synthesis, Structure and Properties. J Clust Sci 33, 1677–1684 (2022). https://doi.org/10.1007/s10876-021-02065-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10876-021-02065-w