
Abstract
Two new borates, [NH3(CH2)3N(CH2)4N(CH2)3NH3][B5O6(OH)4]2 (1) and [NH2(CH2)4N(CH2)2NH3][B7O10(OH)3] (2), have been made under hydrothermal conditions and characterized by infrared spectroscopy, thermogravimetric analysis, powder X-ray diffraction, and single-crystal X-ray structural analysis, respectively. Crystal data for 1: monoclinic, P21/c, a = 9.504(3) Å, b = 14.305(4) Å, c = 10.513(3) Å, β = 90.313(5)°, Z = 2. Crystal data for 2: triclinic, P \( \bar{1}\), a = 9.196(3) Å, b = 9.722(3) Å, c = 11.361(3) Å, α = 71.232(11)°, β = 71.603(10)°, γ = 72.354(12)°, Z = 2. The protonated organic amines are filled in the free space of the hydrogen- bonded network and interact with the inorganic framework by extensive hydrogen bonds.
Graphical Abstract
Two new borates, [NH3(CH2)3N(CH2)4N(CH2)3NH3][B5O6(OH)4]2 (1) and [NH2(CH2)4N(CH2)2NH3][B7O10(OH)3] (2), have been made under hydrothermal conditions. In the structures, the borate anions linked each other via the H-bonding interactions to form the supramolecular inorganic network/frameworks, which further interact with the guest cations through H-bonding and electrostatic interactions, resulting in inorganic–organic hybrid supramolecular frameworks.

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Borate materials have attracted a great deal of attention in the past decades owing to their rich structural chemistry and promising applications in mineralogy, luminescence and nonlinear optical properties [1–6]. From the structural point of view, boron atoms may form strong bonds with oxygen atoms in trigonal planar BO3 and tetrahedral BO4 groups. These groups may further link together by sharing common oxygen atoms to form isolated rings and cages or polymerize into infinite chains, sheets, and frameworks [7–14]. Burns et al. [2, 3, 7] have developed a comprehensive description based on fundamental building blocks (FBBs) to have a clearer nomenclature for the borates with more complicated borate anions.
Up to now, borate materials with various alkali metal, alkaline earth metal, rare earth and transition metal, usually made under high temperature/pressure solid-state conditions, have been covered thoroughly. However, very few information is available regarding the borate system templated by organic amines. Until now, only a few examples with polyanions such as [B4O5(OH)4]2− [15], [B5O6(OH)4]− [16], [B7O9(OH)5]2− [17], [B9O12(OH)6]3− [18] and [B14O20(OH)6]4− [19] have been reported. Therefore, the successful isolation of the larger borate polyanions is highly significant. Herein, we report two new borates, [NH3(CH2)3N(CH2)4N(CH2)3NH3][B5O6(OH)4]2 (1) and [NH2(CH2)4N(CH2)2NH3][B7O10(OH)3] (2), which are made under hydrothermal conditions.
Experimental Section
Materials and Methods
All chemicals used during the course of this work were of reagent grade and used as received from commercial sources without further purification. Elemental analyses of C, H and N were carried out with a Vario EL III elemental analyzer. Infrared (IR) spectra were recorded in the range 400–4,000 cm−1 on an ABB Bomem MB102 spectrometer in KBr pellets.
Synthesis of [NH3(CH2)3N(CH2)4N(CH2)3NH3][B5O6(OH)4]2 (1)
A mixture of H3BO3 (0.240 g), Al(i-PrO)3 (0.160 g), 1,4-Bis(3-aminopropyl)piperazine (0.5 mL), H2O (0.5 mL), and pyridine (4 mL) were stirred at room temperature for 30 min (pH 11.5) and transferred into a 30 mL Teflon-lined autoclave and heated under autogenous pressure at 180 °C for 4 days, and then cooled to room temperature naturally in the air. Colorless bulk crystals were obtained by filtration, washed with distilled water and dried in air (yield: 83 % based on H3BO3). Anal. Calcd for 1, C10H34B10N4O20: C 19.00, H 5.00, N 8.91; Found: C 18.86, H 5.03, N 8.81.
Synthesis of [NH2(CH2)4N(CH2)2NH3][B7O10(OH)3] (2)
A mixture of H3BO3 (0.120 g), NH4B5O8·4H2O (0.120 g), Al(i-PrO)3 (0.160 g), H2O (0.5 mL), N-(2-aminoethyl)piperazine (1 mL) and pyridine (4 mL) were stirred at room temperature for 30 min (pH 11.7) and transferred into a 30 mL Teflon-lined autoclave and heated under autogenous pressure at 180 °C for 5 days, and then cooled to room temperature naturally in the air. Colorless bulk crystals were obtained by filtration (yield: 65 % based on boron), washed with distilled water and dried in air. Anal. Calcd for 2, C6H20B7N3O13: C 17.24, H 4.79, N 10.06; Found: C 17.00, H 5.40, N 9.50.
X-ray Crystallography
The crystal structures of 1 and 2 were determined from single-crystal X-ray diffraction data. The intensity data of 1 and 2 were collected with a Saturn 724 diffractometer using graphite monochromatized Mo Kα radiation (λ = 0.71073 Å) at 293(2) K. Their structures were solved by direct methods and refined by the full-matrix least-squares method on F 2 using SHELXL-97 [20–22]. Intensity data were corrected for Lorentz and polarization effects as well as for an empirical absorption. Experimental details for the structural determinations for 1 and 2 are presented in Table 1. Crystallographic data for the structural analysis has been deposited with the Cambridge Crystallographic Data Centre, CCDC-909682 for 1 and CCDC-909681 for 2.
Results and Discussion
Description of Crystal Structures
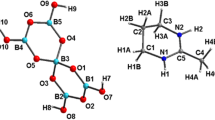
The asymmetric units of compounds 1 (Fig. 1a) and 2 (Fig. 1b) contain 22 and 29 independent non-hydrogen atoms respectively. In the structure 1, the [B5O6(OH)4]− anion consists of two {B3O3} units with three-membered rings (3-MRs) in which four trigonal BO2(OH) (B1, B3, B4 and B5) units and one tetrahedral BO4 (B2) unit are linked by common oxygen atoms (Figs. 1a, 2a). In the triangular BO2(OH) groups, the B–O bonds vary from 1.350 to 1.390 Å and the mean B–O distance is 1.365 Å. In the tetrahedral BO4 groups, the B–O bonds vary from 1.455 to 1.478 Å and the mean B–O distance is 1.468 Å. In 2, however, the large [B14O20(OH)6]4− polyborate anion has been successfully isolated (Figs. 2b, 3), which is formally made by two [B7O11(OH)3]4− (Figs. 1b, 3) units linked by exocyclic oxygen (O3 and O3A) atoms (Fig. 3). Each [B7O11(OH)3]4− cluster unit consists of three 3-MRs in which five trigonal units [three B(1,3,7)O2(OH) and two B(2,4)O3] and two tetrahedral B(5,6)O4 units are linked by common oxygen atoms. The triangularly coordinated boron atoms have B–O distances in the range 1.329–1.386 Å [av. = 1.367 Å] and the tetrahedral B atoms have longer B–O distances in the range 1.433–1.499 Å [av. = 1.471 Å].

The asymmetric units of 1 (a) and 2 (b). A: −x, 1 − y, −z

Polyhedral representation of the FBBs of [B5O6(OH)4]− (a), and [B14O20(OH)6]4− (b) in 1 and 2, respectively

The [B14O20(OH)6]4− cluster in 2 is formally composed of two heptaborate anions, [B7O11(OH)3]4− unit
It is worthy to note that the isolated boron oxoanions having more than six boron atoms are rare [7, 17–19, 23], though many examples containing one to six boron atoms exist in both mineral and synthetic borates [10]. Therefore, the successful isolation of the larger borate polyanion is highly significant. In accordance with the classification scheme of Christ and Clark [5, 24], the shorthand notation for [B5O6(OH)4]− in 1 and [B14O20(OH)6]4− in 2 can be written as ([5]: 4∆ + 1T, isolated) and ([14]: 10∆ + 4T, isolated), respectively.
As expected to be a good building block for supramolecular hydrogen bonded structure, each FBBs, [B5O6(OH)4]− and [B14O20(OH)6]4−, connects each other to form the H-bonding networks by –OH groups oriented in a planar triangular fashion, respectively. In 1, the [B5O6(OH)4]− groups are held together via the O–H···O H-bonding interactions (Table 2), forming 2-D supramolecular network (Fig. 4). The diprotonated [NH3(CH2)3N(CH2)4N(CH2)3NH3]2+ amines are located at the void space of inorganic borate network to compensate the negative charges (Fig. 5a), and further use five H atoms to form N–H···O H-bonding interactions with O atoms of the borate network (Fig. 5b). Similarly, the [B14O20(OH)6]4− clusters in 2 are linked together by the extensive H-bonding interactions to form the interesting 3-D supramolecular framework with regular channels in three directions (Figs. 6, 7a).

Polyhedral view of the hydrogen bonded architectures of 1. Organic amines are not shown for clarity

a View of the H-bonding network in 1, showing the intermolecular H-bonding interactions of the B–O clusters and the intramolecular H-bonding interactions in the protonated amines. The H-bonding interactions between the B–O clusters and the protonated amines are shown as (b)

Polyhedral view of the H-bonding architectures of 2 in the [100] direction

a Polyhedral view of the H-bonding framework of 2, showing the H-bonding interactions between the B–O cluster units. The H-bonding interactions between the B–O cluster units and the protonated amines are shown in (b)
The diprotonated organic molecules, [NH2(CH2)4N(CH2)2NH3]2+, as the guests reside in the channels and interact with the inorganic framework through N–H···O H-bonding interactions (Fig. 7b). The details of H-bonds in 2 are given in Table 3. Interestingly, the H-bonding network in 1 is different from those found in a series of nonmetal borates containing same [B5O6(OH)4]− cluster units, [H3N(CH2)nNH3][B5O6(OH)4]2 (n = 5–6, 8–12) [25], in which the guests with different shapes and sizes result in different H-bonding architectures with circular and rectangulate channels, respectively. These results not only reveal the H-bonding interactions between the borate hosts and the cationic guests, but also show the sizes and shapes of the guest cations may influence the arrangements of inorganic supramolecular networks/frameworks via the H-bonding interactions.
Infrared Spectra
IR spectroscopy was used to verify the nature of the borate groups and the presence of amine in the structure (Fig. 8). For 1, the strong bands at ~1,400, 1,302 and 1,185 cm−1 in the spectrum are consistent with the existence of trigonally coordinated boron, while the bands at 1,110, 1,014, 918 and 886 cm−1 are characteristic of tetrahedral boron. The broad bands in the range of 3,433–3,016 cm−1 correspond to the stretching bands of the N–H, C–H, and O–H groups. The bending bands of N–H and C–H are presented at about 1,639–1,494 cm−1. For 2, the bands at ~1,387, 1,281, 1,185 cm−1 in the spectrum are consistent with the existence of trigonally coordinated boron, while the bands at 1,068–811 cm−1 are characteristic of tetrahedral boron. The broad bands at 3,508–2,835 cm−1 correspond to the stretching bands of the N–H and C–H. The bending bands of N–H and C–H are presented at about 1,606 cm−1.

IR spectra of 1 and 2, respectively
Thermal Properties
As shown in Fig. 9, the TG was carried out in flowing dry air atmosphere with a heating rate of 10 °C·min−1 in the range of 30–1,000 °C. Compound 1 shows two continuous stages of weight

TG curve of 1 and 2, respectively
loss occurring in temperature range of 246–703 °C. The first weight loss is 31.5 % from 246 to 455 °C, assigned to the release of organic amines (calcd 31.7 %), and followed by the loss of 11.3 %, corresponding to the dehydration process of hydroxyls from 455 to 703 °C (calcd 11.2 %). Compound 2 shows continuous stage of weight losses (43.8 %) occurring in range of 30–772 °C, assigned to the release of organic amines and the dehydration process of hydroxyls (calcd 44.3 %), respectively.
Powder X-ray Diffractions
As shown in Fig. 10, the simulated and experimental powder X-ray diffraction (PXRD) patterns, for 1 and 2, are in good agreement in position, indicating the phase purity of the as-synthesized samples of the two compounds.

Simulated and experimental powder XRD patterns of 1 (a) and 2 (b)
Conclusions
In conclusion, the syntheses, crystal structures, and thermal properties of two new organically templated borates, [NH3(CH2)3N(CH2)4N(CH2)3NH3][B5O6(OH)4]2 (1) and [NH2(CH2)4N(CH2)2NH3][B7O10(OH)3] (2), have been made under hydrothermal conditions. The organic guests, accommodated in the free space of H-bonding network, compensate the negative charges and interact with the inorganic framework through strong H-bonding interaction. Considering many kinds of organic guests that can be chosen as the templating agents, it is expected that more borate materials with novel features will be made in future. Further work on this subject is in progress.
References
P. Becker (1998). Adv. Mater. 10, 979.
P. C. Burns (1995). Can. Miner. 33, 1167.
P. C. Burns, J. D. Grice, and F. C. Hawthorne (1995). Can. Miner. 33, 1131.
C. T. Chen, Y. B. Wang, B. C. Wu, K. C. Wu, W. L. Zeng, and L. H. Yu (1995). Nature 373, 322.
C. L. Christ and J. R. Clark (1977). Phys. Chem. Miner. 2, 59.
M. S. Wang, G. C. Guo, W. T. Chen, G. Xu, W. W. Zhou, K. J. Wu, and J. S. Huang (2007). Angew. Chem. Int. Ed. 46, 3909.
J. D. Grice, P. C. Burns, and F. C. Hawthorne (1999). Can. Miner. 37, 731.
C. Y. Pan, G. M. Wang, S. T. Zheng, and G. Y. Yang (2007). J. Solid State Chem. 180, 1553.
J. L. C. Rowsell, N. J. Taylor, and L. F. Nazar (2002). J. Am. Chem. Soc. 124, 6522.
M. Touboul, N. Penin, and G. Nowogrocki (2003). Solid State Sci. 5, 1327.
Z. T. Yu, Z. Shi, Y. S. Jiang, H. M. Yuan, and J. S. Chen (2002). Chem. Mater. 14, 1314.
H. H. Y. Sung, M. M. Wu, and I. D. Williams (2000). Inorg. Chem. Commun. 3, 401.
M. Touboul, N. Penin, and G. Nowogrocki (1999). J. Solid State Chem. 143, 260.
D. M. Schubert, F. Alam, M. Z. Visi, and C. B. Knobler (2003). Chem. Mater. 15, 866.
C. J. Carmalt, W. Clegg, A. H. Cowley, F. J. Lawlor, T. B. Marder, N. C. Norman, C. R. Rice, O. J. Sandoval, and A. J. Scott (1997). Polyhedron 16, 2325.
Q. Li and T. C. W. Mak (1997). Supramol. Chem. 8, 147.
Z. H. Liu and L. Q. Li (2006). Cryst. Growth Des. 6, 1247.
D. M. Schubert, M. Z. Visi, and C. B. Knobler (2000). Inorg. Chem. 39, 2250.
Z. H. Liu, L. Q. Li, and W. J. Zhang (2006). Inorg. Chem. 45, 1430.
G. M. Sheldrick. SHELXTL 97, Program for Crystal Structure Refinements (University of Göttingen, Göttingen, 1997).
G. M. Sheldrick. SHELXS97, Program for Crystal Structure Solution (University of Göttingen, Göttingen, 1997).
G. M. Sheldrick. SADABS Program for Siemens Area Detector Absorption Corrections (University of Göttingen, Göttingen, 1997).
C. Y. Pan, G. M. Wang, S. T. Zheng, and G. Y. Yang (2007). Z. Anorg. Allg. Chem. 633, 336.
G. Heller (1986). Top. Curr. Chem. 131, 39.
M. Z. Visi, C. B. Knobler, J. J. Owen, M. I. Khan, and D. M. Schubert (2006). Cryst. Growth Des. 6, 538.
Acknowledgments
The authors are thankful for the financial supports from the National Nature Science Fund (NNSF) of China (Nos. 91122028, 21221001 and 50872133), the NNSF for Distinguished Young Scholars of China (No. 20725101), and 973 Program (No. 2011CB932504).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wu, LZ., Cheng, L. & Yang, GY. Syntheses and Structures of Two New Borates: [NH3(CH2)3N(CH2)4N(CH2)3NH3][B5O6(OH)4]2 and [NH2(CH2)4N(CH2)2NH3][B7O10(OH)3]. J Clust Sci 24, 811–820 (2013). https://doi.org/10.1007/s10876-013-0577-0
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10876-013-0577-0