
Abstract
Synthesis, characterization and structural relevance of four new copper carboxylate complexes (1–4) has been presented here. The complexes have been synthesized by direct treatment of the substituted phenyl acetate and pyridine ligands in aqueous medium. The complexes were stable indefinitely with excellent yields and were characterized using spectroscopic and single crystal XRD techniques. FTIR spectroscopy revealed the bridging bidentate coordination mode for the carboxylate moiety in accordance to the actual structure revealed by XRD. Moreover, UV-Visible spectroscopic and cyclic voltammetric studies helped in their characterization and yielded signals which were typical of the copper(II) complexes. Successfully solved single crystal XRD data showed binuclear paddlewheel structures for all the complexes with both copper ions linked through four OCO bridges of ortho-methoxy phenyl acetate (1–3) and ortho-methyl-meta-nitrophenyl acetate (4). The geometry around each copper was distorted square pyramidal where the apical positions are occupied by meta-bromopyridine (1), meta-methylpyridine (2) and DMSO (3 and 4) molecules. The complexes exhibited excellent DNA-binding activity majorly via intercalation as revealed by four experimental techniques. This preliminary study showed that the synthesized complexes add to the existing treasury on the paddlewheel complexes.
The Index Abstract
Air stable newly synthesized and purified crystalline complexes with their preliminary structural relevance.

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Exploration of new copper complexes with new structural features and improved properties is underway [1,2,3]. Paddlewheel copper complexes constitute an important class of copper carboxylate complexes. In such complexes two copper ions are bonded by four carboxylate bridges thus a stable unit containing two doubly charged copper(II) ions is resulted. These four carboxylate bridges constitute the square base of the square pyramid which is resulted when another mono-dentate ligand is attached to each copper ion [4, 5]. Thus each copper ion is penta-coordinated in a paddlewheel structure with square pyramidal geometry. However, owing to the geometric restrain offered by four carboxylate bridges between the two copper ions in a paddlewheel structure, both ions are forced to lie closer to one another. That is why the inter-atomic distance between copper ions within the paddlewheel is mimicked as a bond by a crystallography program and thus a bond is drawn by the software between the two ions. However, this is not a genuine bond because no orbital on penta-coordinated copper(II) ion is oriented towards the other copper(II) ion to result in a substantial overlap. Rather, the dx2-y2 orbital containing the unpaired electron is oriented towards the carboxylate bridges in the square base rather than the other copper ion in the paddlewheel unit. Thus the orbitals are oriented away from one another where no overlap is possible [6, 7].
Paddlewheel complexes are found in several structural types. If the apical ligand is trans-bidentate, it is able to bind two paddlewheel subunits at its both ends. Thus the apical ligand acts as a linker between the adjacent paddlewheel subunits and the resulting structure is a one dimensional infinite array of the paddle wheel units [8,9,10]. In another type, the subunits are interlinked through the linker units present on the carboxylate ligands as well. Such complexes result in a two dimensional sheet structures [11], three dimentional structures and padllewheel based MOFs [12]. There is another type where the subunits are interlinked by their own Cu- and O-atoms without the intervention of a linker apical ligand. In these polymeric complexes, the paddlewheel subunits are directly interconnected resulting in a stepped polymeric structure [13]. Despite the aforementioned types, the most common type is the one which has discrete paddlewheel units in its structure [14]. These have relatively small sized ligands, often a single atom such as Cl-, Br- at the apical position. Complexes of the last type has better solution properties, solubility and stability and thus widespread applications. We have synthesized new complexes of the same type derived from substituted phenyl acetate ligands and N-donor apical ligands. Their structures have been correlated mutually as well as with literature along with exploration of some other structure related properties.
Experimental
Materials and Methods
All the solvents and reagents were of analytical grade. Distilled water was used throughout the study for synthesis and spectroscopic studies. Substituted derivatives of phenyl acetic acid were purchased from Fluka. FTIR spectra were recorded on a Nicolet-6700 FTIR spectrometer equipped with attenuated total reflectance technique in the range 4000 –400 cm− 1. DNA interaction studies were performed on a Beckman U-2020 UV–Visible spectrophotometer and Ubbelohde viscometer. For spectrofluorimetry, a PerkinElmer LS 45 fluorescence spectrometer with slit width of 10 nm was employed. Cyclic voltammetry was performed using SP-300 potentiostat with a typical three electrode cell.
Single Crystal X-ray Analyses
Crystallographic data of the complexes were acquired using KAPPA APEX-II CCD diffractometer. The diffractometer was equipped with graphite monochromatic radiations Mo-Kα (λ = 0.71073 Å). SAINT and multi-scan [15] were employed for data reduction and absorption corrections. Structures were solved by SHELXT-2014 [16] and refined within the WinGX package with SHELXL-2019/2 [17].
Moreover, dimethylsulfoxide (DMSO) was disordered over three positions with occupancy ratio 0.786(4): 0.132(4): 0.082(4) in solvates of complexes 1 and 2 (not the DMSO ligands) while the nitro-groups were disordered over two positions in complex 1. The disorder was solved by using various restraints. The thermal parameters of the atoms in both parts of the disordered nitro-groups were made equal to each other by using “EADP” restraint. The nitro-groups were made flat by using “FLAT” restraint. For stabilization of the disordered parts of DMSO, thermal parameters of part 1 are made equal to each other. Similar procedure was done for other disordered parts of DMSO. DFIX and DANG restraints were used for making bond lengths and bond angles of the disordered parts in a usually acceptable range.
Experimental Protocols of DNA Interaction Studies
For absorption spectroscopy, concentrations of the complexes were optimized at 10 mM and absorbance of the pure complex solutions were taken in DMSO: water system (4:1) and then in the presence of 10, 20, 30, 40, 50, 60, 70 and 80 µM DNA.
For cyclic voltammetry, the compounds were dissolved in DMSO at 4.5 mM and KCl was added as electrolyte. Voltammograms of these sample solutions were recorded in pure form and then in the presence of 15, 25, 35, 45, 55, 65, 75 and 85 µM DNA.
For DNA-binding study through spectrofluorimetry, complex and ethidium bromide mixture (1:1) at 10− 6 M each was prepared in phosphate buffer (pH = 7.2). Emission spectra of the mixture were taken in pure form and in the presence of 10, 20, 30, 40, 50, 60, 70 and 80 µM DNA.
DNA-binding study through viscometry was performed by running the aqueous DNA solution (5 × 10− 5 M) in the Ubbelohde visometer and the time was noted by stopwatch. Then 10, 20, 30, 40, 50 and 60 µL solutions (5 × 10− 3 M) of each of the complexes was added to the same volume of DNA solution and its time of running in viscometer was noted each time. The relative viscosity was calculated from the difference in time of running.
Syntheses of the Complexes
Complexes 3 and 4 were prepared by treating the aqueous sodium salts of ortho-methoxyphenyl acetate (0.84 g, 5 mmol) with copper sulphate (0.622 g, 2.5 mmol) and stirred for 3 h at 60 oC. Then 3-bromopyridine (1) (0.24 mL, 2.5 mmol) or 3-methylpyridine (2) (0.24 mL, 2.5 mmol) was added and stirred for 3 h again. The final products w ere the solid precipitates which were washed with distilled water and air dried as shown in scheme 1. Purification and final crystallization from methanol yielded 3 and 4. Recrystallization of the dried sample of 3 from DMSO yielded 2 as shown in scheme 2. Recrystallization of the dried sample of an already published compound [18] by our research group yielded fine crystals of 1 as shown in scheme 3. These crystals were analyzed by single crystal XRD technique.
Complex 1: Green crystals; m.p. 173–175 °C; yield (60%). FT-IR (cm− 1): 1621 ν(OCO)asym, 1421 ν(OCO)sym, ∆ν = 200, 3058 ν(Ar-H), 2972 ν(CH2), 1610, 1459 Ar(C = C), 1242 ν(O-CH3), 482 ν(Cu-O).
Complex 2: Light blue; m.p. 179–180 °C; yield (68%). FT-IR (cm− 1): 1604 ν(OCO)asym, 1415 ν(OCO)sym, ∆ν = 189, 3078 ν(Ar-H), 2956 ν(CH2), 1604, 1465 νAr(C = C), 1246 ν(O-CH3), 493 ν(Cu-O).
Complex 3: Light blue; m.p. 162–163 °C; yield (70%). FT-IR (cm− 1): 1663 ν(OCO)asym, 1463 ν(OCO)sym, ∆ν = 200, 3074 ν(Ar-H), 2939 ν(CH2), 1585, 1463 νAr(C = C), 1247 ν(O-CH3), 428 ν(Cu-N), 503 ν(Cu-O).
Complex 4: Green; m.p. 152–153 °C; yield (75%). FT-IR (cm− 1): 1644 ν(OCO)asym, 1428 ν(OCO)sym, ∆ν = 216, 3040 ν(Ar-H), 2925 ν(CH2), 1589, 1420 ν(C = C), 1455, 1349 ν(NO2), 420 ν(Cu-N), 508 ν(Cu-O).
Results and Discussion
Four new copper complexes have been synthesized and isolated quantitatively following schemes 1, 2 and 3. These have been obtained in crystallized form. These have been structurally characterized and the spectroscopic results are in harmony with structural elucidation results. Moreover, the complexes have been subjected to their DNA-binding assay.

synthesis of complexes 1 and 2

Preparation of complex 1

Preparation of complex 2
FTIR Study
FTIR spectroscopy of the pure samples of the complexes revealed all the peaks which were expected in the structures of the synthesized complexes. The most prominent peaks were those of the stretching vibrations of the carboxylate moiety. The asymmetric stretch of the carboxylate moieties of all the complexes were observed in the range 1621 –1544 cm− 1 while the symmetric stretching vibrations were in the range 1465 –1428 cm− 1. The difference between the two peaks ∆ν for complexes 1–4 was 200, 189, 200 and 216 cm− 1, respectively. These ∆ν values were in the range (150–250 cm− 1) typical of bridging bidentate coordination mode of the carboxylate moiety [19]. When the ∆ν values are below 150 cm− 1, it shows a chelate bidenatate coordination mode while ∆ν values above 250 cm− 1 has been found to indicate monodentate or uncoordinated carboxylate moiety [19]. Thus for metal carboxylate complexes, ∆ν value can give valuable clue regarding the nature of carboxylate moiety.
Aromatic C-H was observed on 3058, 3078, 3074 and 3040 cm− 1, respectively for complexes 1–4. A closely lying peak was that of methylene CH2 which was observed just below 3000 cm− 1 for all the complexes. Similarly, ligand to copper bond was indicated by Cu-O bond stretching observed at 482, 493, 503 and 508 cm− 1, respectively for complexes 1–4 as observed for the same functionality in other structurally characterized copper(II) complexes [20, 21]. The aromatic C = C bond was also indicated by its respective peaks. The FTIR spectra were in agreement to the structural data of the complexes.
Structural Description
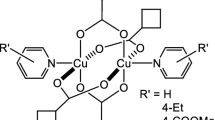
Molecular structures of complexes 1–4, drawn in mercury have been shown in Fig. 1 (A-D) while their crystallographic parameters have been listed in Tables 1 and 2. The complexes crystallized in triclinic crystal system with space group P-1. Each complex is dinuclear with two copper ions bonded by four carboxylate ligands in syn-syn fashion. These four Cu-O bonds around each copper ion constitute the square base of the square pyramidal geometry around each copper. The apical position of the square pyramid is formed by the oxygen atom of the DMSO molecule in complexes 1 and 2 and by the nitrogen atom of 3-methylpyridine and 3-bromopyridine in 3 and 4, respectively.
The paddlewheel complexes of copper(II) have both copper ions drawn closer enough by the four OCO bridges so that the inter-nuclear distance between the two ions is comparable to a covalent bond. However, a bond cannot be formed between the two copper ions along that axis because there is no suitably oriented orbital on each copper to overlap and form a bond along that axis. The central Cu-Cu bond in these molecules is not a genuine bond, rather a bond drawn by the software owing to the shorter distance between two copper ions. A crystallography software/program usually draws a bond between such ions owing to the short distance but that bond is not meaningful and metal ions are actually 5-coordinated square pyramidal in these complexes.

Molecular structures of complexes 1–4 (A-D) drawn at 50% probability ellipsoids in mercury. H atoms have been omitted for clarity
The metal ion is bulging out of the square base towards the apical position in such complexes. It means that the Cu···Cu ̶ O angle is less than 90o and the O ̶ Cu ̶ O/Napical is greater than 90o. The distance between two Cu ions within a paddlewheel is short owing to the four carboxylate bridges between the two ions. However, no orbital on these Cu ions is oriented suitably to allow an overlap between the two ions so no genuine bonding interaction can occur between these Cu ions. However, this Cu···Cu distance in paddlewheel complexes is sensitive to the basic strength of the carboxylate ligand. Thus varying the basic strength of the donor atoms, this distance can be increased or decreased and the properties dependent on this parameter can be tuned. Changing the carboxylate ligand from trifloroacetato (2.766(1) Å) to trimethylacetato (2.630(18) Å) the Cu···Cu distance is shortened as the basicity of the ligand is increased [22]. The corresponding Cu···Cu distances in the synthesized complexes lie in the range 2.619(5) − 2.671(13) Å. The basic strength of the carboxylate ligands is moderate so Cu···Cu separation is less than that of the trifloroacetate based complexes [22]. The intermolecular interactions have been presented in the packing diagrams of complexes 1–4, shown in Fig. 2A-D.

Packing diagrams of the synthesized complexes 1–4 (A-D) viewed along the axes
Supra-molecular Chemistry
Since there is no O/F/N─H∙∙∙O/F/N interactions in the lattices, there are some C─H∙∙∙O interactions in complex 4. Other weak interactions in the lattices of all the four complexes give rise to interesting molecular arrays as shown in Fig. 2. Since H atoms are towards the periphery of the molecules, the greatest contribution is that of H∙∙∙H interactions followed by C─H∙∙∙π interactions. The latter is owing to the phenyl rings present in structure of each molecule.
Stability Studies in Solution form
In order to ascertain whether the structure and geometry of the synthesized complexes remains intact in solution form, their solutions were subjected to absorption spectroscopic studies at various intervals. Since the absorption spectrum of a copper(II) complex is a function of geometry (octahedral, square planar, square pyramidal, trigonal bipyramidal and tetrahedral) around copper ion, the spectra for solution studies were taken in visible region of spectrum as shown in Fig. 3. These spectra show that the complexes remain unchanged for at least 24 h in common solvents.

Absorption spectra of complexes 1–4 at various time intervals
DNA-Binding Studies of the Complexes
DNA-binding potential of the complexes was explored using four different techniques. These are discussed below in detail one by one.
UV-Visible Spectrophotometry
The complexes gave rise to ligand based well-defined absorption peaks in the UV-region following the Beer-Lambert’s Law. It means that reduction in concentration of the complexes will result in reduction in their absorbance value as per the Beer-Lambert’s Law. Adding incremental quantity of DNA will result the reduction in concentration of the complex owing to the complex-DNA adduct formation. This has been shown for 1–4 in Fig. 4, where the absorbance value of the pure complex represented by the uppermost plot has suffered proportionate diminution on DNA-addition. This indicates that each complex effectively binds with DNA majorly through intercalative mode of binding [23, 24]. Since there is pronounced reduction in absorbance of each complex on addition of DNA and the wave length of complex suffers no appreciable change along the successive addition, such binding mode is called intercalative binding where the complex molecules simply insert between the DNA base pairs [23, 24]. This binding mode is not unexpected because the complexes have plenty planar aromatic moieties in the structure through which these molecules get inserted into the DNA double strand. The binding ability has been quantitatively ascertained by calculating the binding constant Kb using a simplified form of the Benesi-Hildebrand equation [25]. This was done by plotting the reciprocal molar concentration of DNA vs. the relative absorbance value as shown in Fig. 5 and the Kb value was calculated from the slope to intercept ratio of the plot. The Kb values calculated were 1.85, 2.05 and 1.88 and 1.92 × 104 M− 1, respectively for complexes 1–4. Similar DNA binding potency was observed for other copper complexes as well [26].

Absorbance spectra of complexes 1–4 in the absence of DNA (upper most peak designated as a) and presence of 10, 20, 30, 40, 50, 60, 70 and 80 µM DNA (designated as b-i) showing decrease in absorbance with the addition of DNA

Plots of 1/[DNA] vs. Ao/(A-Ao) for the calculation of the binding constants of complexes 1–4
DNA-Binding Study Using Viscometry
The viscosity of DNA is sensitive to its strand length; longer strains have higher viscosity than shorter strands. Planar molecules are able to intercalate between base pairs of DNA double strands which will lengthen the DNA strand resulting in higher viscosity [23, 24]. When the complexes 1–4 were added gradually to the DNA solution, the viscosity of DNA solution increased steadily as seen in the Fig. 6. This also indicated the intercalative mode of complexes with DNA. The same mode was indicated by UV-Visible spectroscopy as well.

Plots of the ratio of molar concentrations of complex/DNA vs. relative viscosity for complexes 1–4
DNA-Binding Study Using Spectrofluorimetry
Ethidium bromide (EtBr) is a potent intercalator into the DNA-base pairs. It has high emission intensity in DNA-bound form while in free state its emission intensity is quenched by the buffered solvent medium. If another molecule competing for intercalation into the DNA-base pairs is added to the solution containing DNA-EtBr adduct, it will compete with EtBr and will tend to replace the later [27]. If the added molecule successfully replaces the EtBr and gets inserted into the DNA strand, the emission intensity of the EtBr will be reduced since the quantity of the DNA-bound and emissive EtBr has been reduced. On successive addition of the synthesized complexes 1–4 to the solution containing DNA-EtBr adduct, its emission intensity was reduced on each addition. This showed that the complexes are able to compete successfully with EtBr for intercalation into the DNA base pairs as shown in Fig. 7 for complexes 1–4. This technique indicated that the complexes are more potent intercalators than EtBr and that the mode of binding of the complexes with DNA is intercalation as deduced from the previous techniques.

Decrease in fluorescence intensity (indicated by the downward headed arrow) of ethidium bromide in the presence of 10, 20, 30, 40, 50, 60, 70 and 80 µM complex added to the solution containing DNA-EtBr adduct. A-D correspond to spectra of complexes 1–4
DNA-Binding Study Using Cyclic Voltammetry
Change in the concentration of an electro-active compound can be followed by cyclic voltammetry where the resulting current is changed with change in concentration. In this context, cyclic voltammograms of the complexes were recorded before and after adding variuos quantities of DNA. The voltammograms did suffer deminution in current on addition of DNA which indicated binding with the complexes as shown in Fig. 8. The diminution in current of the complexes on addition of DNA was used to calculate the binding constant of the complexes with DNA using plots shown in Fig. 9. The results of the four techniques on DNA-binding study are in harmony with each other.

Cyclic voltammograms of complexes 1–4 (A-D) in the absence of DNA (upper most voltamogram designated as a) and presence of 15, 25, 35 and 45 µM DNA (designated as b-e) showing decrease in absorbance with the addition of DNA

Plots of the log. of current vs. log. of 1/DNA of complexes 1–4
Conclusion
Four new carboxylate complexes of copper(II) (1–4) have been synthesized with substituted phenyl acetic acids and pyridine. These were characterized using UV-Visible and FTIR spectroscopy and single crystal XRD. The FTIR study showed the bands which indicated their synthesis and attachment of ligands to copper ion. The complexes have been explored for the DNA-binding activity studied by UV-Visible and fluorescence spectroscopy, cyclic voltammetry and viscometry. All the four techniques yielded coherent results of the activity. This indicated facile and potent DNA-binding activity of the synthesized complexes. This preliminary study showed that the synthesized complexes can add to the existing treasury on the paddlewheel complexes.
Data Availability
No datasets were generated or analysed during the current study.
References
Vakulka A, Goreshnik E (2024) Copper(II) Tetrafluoroborate Hexahydrate: Preparation, structure and Raman Spectrum. J Chem Crystallogr. https://doi.org/10.1007/s10870-024-01008-3
Cherutoi JK, Fronczek FR, Maverick AW (2023) Triphenylamine-based Bis(β-Diketones) for construction of Supramolecular copper(II) complexes. J Chem Crystallogr 53:475–482. https://doi.org/10.1007/s10870-023-00986-0
Cukierman DS, Ligiero CBP, de Avillez RR et al (2023) Crystal Structure and Hirshfeld Analysis of a Poorly Water Soluble Bis(ligand)copper(II) Complex containing the Metallophore Pyridine-2-Carboxaldehyde 2-Furoyl Hydrazone. J Chem Crystallogr 53:8–15. https://doi.org/10.1007/s10870-022-00955-z
Wang Z, Huang Y, Xu K et al (2023) Natural oxidase-mimicking copper-organic frameworks for targeted identification of ascorbate in sensitive sweat sensing. Nat Commun 14, 69 (2023). https://doi.org/10.1038/s41467-022-35721-4
Halcrow MA (2013) Jahn-Teller distortions in transition metal compounds, and their importance in functional molecular and inorganic materials. Chem Soc Rev 42:1784–1795
Iqbal M, Ali S, Tahir MN et al (2020) A binary copper(II) complex having a stepped polymeric structure: synthesis, characterization, DNA-binding and anti-fungal studies. J Serb Chem Soc 85(2):203–214
Li L, Xia SS, Hu XJ et al (2024) A highly porous phosphonocarboxylate metal–organic framework for hydrogen storage. Struct Chem https://doi.org/10.1007/s11224-024-02305-5=7
Peskov MV, Miao X-H et al (2013) Electronic and magnetic properties of infinite 1D chains of Paddlewheel carboxylates M2(COOR)4 (M = Mo, W, Ru, Rh, ir, Cu). J Phys Chem C 117(10):5462–5469
Marie L, Mortensen, Joanna N, Vo, Maxwell U, Hyder A, Shrivasta GT, McCandless KJ, Balkus (2023) Transformation of a copper complex to a 1-D coordination polymer to 3-D metal-organic frameworks. Polyhedron 242:116512
Kosaka W, Zhang J, Watanabe Y et al (2022) Considerations on gated CO2 adsorption behavior in one-dimensional porous coordination polymers based on paddlewheel-type Dimetal complexes: what determines gate-opening temperatures? Inorg Chem 61:32, 12698–12707
Schnabel J, Schulz A, Lunkenheimer P et al (2023) Benzothiadiazole-based rotation and possible antipolar order in carboxylate-based metal-organic frameworks. Commun Chem 6, 161 (2023). https://doi.org/10.1038/s42004-023-00959-6
Shi X, Zu Y, Li X et al (2023) Highly selective adsorption of light hydrocarbons in a HKUST-like MOF constructed from spirobifluorene-based octacarboxylate ligand by a substitution strategy. Nano Res 16:10652–10659
Iqbal M, Ali S, Tahir MN et al (2019) Mono- and poly-nuclear copper(II) carboxylates with flourous ligands: synthesis, structure and improved properties. Inorg Chim Acta 498:119177
Chakraborty P, Chatterjee A, Mandal R et al (2023) Synthesis, crystal structure, and catechol oxidase activity of a di-nuclear paddle-wheel Cu (II) carboxylate complex. Struct Chem. https://doi.org/10.1007/s11224-023-02225-w
Bruker (2005) APEX2, SAINT and SADABS. Bruker AXS Inc., Madison, Wisconsin, USA
Sheldrick GM (2015) SHELXT–Integrated space-group and crystal-structure determination. Acta Crystallogr A: Found Adv 71:3–8
Sheldrick GM (2015) Crystal structure refinement with SHELXL. Acta Cryst C 71:3–8
Mushtaq A, Ali S, Iqbal M, Tahir MN, Ismail H (2017) Synthesis of a New Heteroleptic copper(II) complex: structural elucidation, DNA binding and In-vitroAlpha glucosidase inhibition studies. J Chem Soc Pak 39:471–477
Iqbal M, Ahmad A, Ali S et al (2013) Dimeric ‘‘paddle-wheel’’ carboxylates of copper(II): synthesis, crystal structure and electrochemical studies. Polyhedron 50:524–531
Dakua VK et al (2023) Synthesis, crystal structure, Hirshfeld surface, and DFT studies of a copper(II) complex of 5,5′-dimethyl-2,2′-bipyridine and 1,2,2-trimethylcyclopentane-1,3-dicarboxylic acid. Results Chem 6:101050
Hussain A, AlAjmi MF, Rehman MT et al (2019) Copper(II) complexes as potential anticancer and nonsteroidal anti-inflammatory agents: in vitro and in vivo studies. Sci Rep 9:5237
Iqbal M, Ali S, Muhammad N, Sohail M (2013) Synthesis, crystal structures and electrochemical characterization of dinuclear paddlewheel copper(II) carboxylates. Polyhedron 57:83–93
Kolbeck PJ, Vanderlinden W, Gemmecker G et al (2021) Molecular structure, DNA binding mode, photophysical properties and recommendations for use of SYBR Gold. Nucleic Acids Res 49:5143–5158
Berdnikova DV, Sosnin NI, Fedorova OA, Ihmels H (2017) Governing the DNA-binding mode of styryl dyes by the length of their alkyl substituents – from intercalation to major groove binding. Org Biomol Chem 16:545–554
Benesi HA, Hildebrand JH (1949) A Spectrophotometric Investigation of the Interaction of Iodine with aromatic hydrocarbons. J Am Chem Soc 71:2703–2707
Sureshbabu P, Varghese B, Sujitha E, Sabiah S (2022) Syntheses, structure, DNA docking and antimicrobial studies of copper(II) complexes with diethylenetriamine and N-bidentate ligands. Inorg Chim Acta 536: article 120898
Phadte AA, Banerjee S, Mate NA, Banerjee A (2019) Spectroscopic and viscometric determination of DNA-binding modes of some bioactive dibenzodioxins and phenazines. Biochem Biophys Rep 18: article 100629
Acknowledgements
NA.
Author information
Authors and Affiliations
Contributions
The funds were received and work was supervised by Saqib Ali. Work was done and manuscript prepared by Muhammad Iqbal and Afifa Mushtaq. The activities were performed by Khadija Shahid. The crystallographic work was done by Muhammad Nawaz Tahir.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Conflict of interest
The authors claim no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary material
Supplementary material
Crystallographic data for the complexes 1–4 reported in this paper have been deposited with the Cambridge Crystallographic Data Centre corresponding to CCDC #s 2,068,200, 2,068,201, 2,068,198 and 2,068,197, respectively. [Fax: +44 (1223)336 033]; e-mail: deposit@ccdc.cam.ac.uk.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Mushtaq, A., Iqbal, M., Rashid, Z. et al. Synthesis, Structural Elucidation and DNA-Binding Studies of Centrosymmetric Paddlewheel Copper Carboxylate Complexes. J Chem Crystallogr 54, 261–273 (2024). https://doi.org/10.1007/s10870-024-01018-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10870-024-01018-1