
Abstract
The P2X7 receptor (P2X7R) is an ion channel which is activated by interactions with the extracellular ATP molecules. The molecular complex P2X7R/ATP induces conformational changes in the protein subunits, opening a pore in the ion channel macromolecular structure. Currently, the P2X7R has been studied as a potential therapeutic target of anti-inflammatory drugs. Based on this, a series of eight boronic acids (NO) analogs were evaluated on the biologic effect of this pharmacophoric group on the human and murine P2X7R. The boronic acids derivatives NO-01 and NO-12 inhibited in vitro human and murine P2X7R function. These analogs compounds showed effect better than compound BBG and similar to inhibitor A740003 for inhibiting dye uptake, in vitro IL-1β release and ATP-induced paw edema in vivo. In both, in vitro and in vivo assays the compound NO-01 showed to be the hit compound in the present series of the arylboronic acids analogs. The molecular docking suggests that the NO derivatives bind into the upper body domain of the P2X7 pore and that the main intermolecular interaction with the two most active NO derivatives occur with the residues Phe 95, 103 and 293 by hydrophobic interactions and with Leu97, Gln98 and Ser101 by hydrogen bonds.. These results indicate that the boronic acid derivative NO-01 shows the lead compound characteristics to be used as a scaffold structure to the development of new P2X7R inhibitors with anti-inflammatory action.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
P2X family takes part in the purinergic signaling network where ATP (adenosine 5′-triphosphate), other nucleotides and nucleosides are the extracellular signaling molecules (Bartlett et al. 2014). Together with P2Y family, P2X receptors are involved in the cellular response to extracellular ATP molecules (Baas 2012). P2X receptors are trimeric ligand-gated cation channels whose family consists of seven members of receptors (P2X1–7). Comparatively, the monomeric unit of the P2X7 receptor (P2X7R) is the highest among members of the P2X family. This subtype also differs from the others because it has a large number of amino acids residues in the intracellular C terminus (Baas 2012; Burnstock and Knight 2018). P2X7R is present in many cell types including hematopoietic lineages, epithelial and endothelial cells, fibroblasts, osteoblasts, astrocytes, Schwann cells, some populations of neurons, and cells from microglia, among others (Bartlett et al. 2014).
Differently from the other members of the P2X family, P2X7R when activated for micromolar ATP concentrations leads to the opening of the channels for the flux of small cations (Na+, K+ or Ca2+) through the plasma membrane. When P2X7R is stimulated for high ATP concentrations (mM range) and, after prolonged or repeated stimulation, thus a larger permeability state is reached forming or inducing a large conductance ionic channel (Faria et al. 2017; North 2002). Molecules with a mass of up 900 Da can permeate the membrane by these large pores, which makes possible the liberation of inflammatory cytokines (Young and Górecki 2018). Activation of P2X7R gives rise to a sequence of signaling events, which are reliant on the cell type expressing this receptor, the ATP extracellular concentration and other conditions of the extracellular medium (Bartlett et al. 2014).
According to the cell type, P2X7R activation modulates different signaling pathways leading to the activation of the caspase-1-containing inflammasome NLRP3, release of proinflammatory molecules; such as interleukin 1β (IL-1β), interleukin-18 (IL-18) and interleukin-36α (IL-36α); activation of metalloproteases and other proteases, the formation of reactive oxygen species (ROS) and other events. There are many evidences which support that the P2X7R is an essential target to be investigated for the treatment of a variety of diseases (Bartlett et al. 2014; Chen et al. 2018; Gorodeski 2012; Savio et al. 2018; Skaper 2011; Sugiyama 2014; Tsuda et al. 2010). Several diseases states are associated with P2X7R single nucleotide polymorphisms (SNPs) (Volonté et al. 2012) and other related to the role that P2X7R exerts in disorders such as the inflammatory and immune response, for instance (Baudelet et al. 2015; Rech et al. 2016). Therefore, the development of P2X7R inhibitors may be a relevant strategy for the discovery of new drugs for the treatment of inflammatory conditions such as rheumatoid arthritis (Mehta et al. 2014).
A conserved tyrosine residue (tyrosine-343) in the carboxyl terminus of the P2X7R, when phosphorylated can affect the function of this receptor (Kim et al. 2001). Taking this information into account, Wiley and co-workers evaluated the influence of 18 protein tyrosine kinase inhibitors on the 86Rb+ efflux mediated by the human P2X7R. A phthalazinamine derivative (compound P), which inhibits the vascular endothelial growth factor receptor kinase, was found to be the most potent compound, blocking the ATP-induced cation efflux by 76% in human B-lymphocytes and by 66% in erythrocytes in a dose-dependent manner (half-maximum inhibitory concentration - IC50 ≅ 5 μM) in both cells. The authors suggested that compounds targeting the ATP-binding sites of kinases would be potential blockers of the P2X7R (Shemon et al. 2006).
An important class of inhibitors of tyrosine kinases is the tyrphostin group which is benzylidene malononitriles molecules that can inhibit the cell-signaling transduction by decreasing tyrosine phosphorylation. Tyrphostins are tyrosine kinase (TYK) inhibitors enzymes that play critical roles in the inflammation process. Therefore, tyrphostins molecules are bioactive compounds which have been shown a promising anti-inflammatory activity (Fig. 1) (Dimitrova and Ivanovska 2013; Gyurkovska et al. 2014).

Anti-inflammatory tyrphostins examples and potential activities for this class of TYKs inhibitors
Recently, the effect of introducing the boronic acid group B(OH)2 in arylcyanovinyl compounds, that can be classified as tyrphostins compounds was investigated in their anticancer properties by our research group. A series of boronic acid derivatives were synthesized, generating the compounds of NO series (Fig. 2). From this boronic acid analogs series, the compound NO-12 displayed high antiproliferative activity against GH3 cell lines from rat pituitary tumor (0.14 ± 0.09 μM), whereas compound NO-13 has shown antiproliferative potential against the human leukemic monocyte lymphoma cell line U937 (9.83 ± 1.07 μM) (Hiller et al. 2018).

NO series synthesized by Knoevenagel condensation of the aromatic aldehydes (Hiller et al. 2018)
In addition to compounds in the NO series, there are other studies showing the introduction of the B(OH)2 unit into the aromatic ring of known anticancer compounds resulting in better activities or bioavailability or modifying the performance profile of these compounds (Ahmed et al. 2006; Asano et al. 2004; Ban et al. 2009; Bradke et al. 2008; Kumar et al. 2003; LeBeau et al. 2008; Modzelewska et al. 2006; Nakamura et al. 2006; Shimizu et al. 2010). Bortezomib, for example, is proteasome inhibitor current in use in multiple myeloma therapy (Buac et al. 2013; Diaz and Yudin 2017; Dou and Zonder 2014). Due to their empty p orbital, boronic acids can coordinate to the heteroatoms present in enzymes and receptors in a reversible covalent mechanism. These characteristics are responsible for the recent growing interest both in the industry and in the academy for the development of new boron-based drugs (Baker et al. 2009; Ban and Nakamura 2015; Das et al. 2013, 2013).
Since arylboronic acids derivatives can show different pharmacological activities, in the present work, the action of NO series compounds was evaluated on the P2X7R. This study was based on the following observations from the literature reports and our group’s results: 1) tyrosine kinase inhibitors may be potential blockers of the P2X7R; 2) tyrphostins are tyrosine kinase inhibitors whose antiinflammatory activity has aready been demonstrated; 3) the introduction of the B(OH)2 moiety into known anticancer agents, including tyrosine kinase inhibitors, led to compounds with improved activity in many cases; 4) tyrphostins containing the B(OH)2 moiety can interact with residues by both hydrogen and covalent bonds; 5). To the best of our knowledge, tyrphostins containing B(OH)2 moiety have not yet been evaluated as blockers of the P2X7R (Groziak 2001; Koehler and Lienhard 1971; Matthews et al. 1975; Philipp and Bender 1971; Suenaga et al. 1996; Weston et al. 1998; Zervosen et al. 2012).
Materials and methods
Chemistry
Materials and apparatus were used for the NO series synthesis. Common and deuterated solvents, ethyl cyanoacetate, malononitrile, and cyanoacetic acid were purchased from Sigma Aldrich Brazil LTDA. Boronic acids were purchased from Combi-Blocks. All these substances were employed as received, without being previously submitted to purification processes. Products (NO series) were purified by recrystallization using a suitable solvent. The reported yields are an average of triplicates and refer to the isolated ones after recrystallization. The melting points (mp) of the pure products were recorded on a Fisatom 413D apparatus. Reaction progress was monitored through analytical thin-layer chromatography (Sillicycle Ultrapure Silica Gels, F254) and the spots were visualized by UV light or by reaction with 2,4-dinitrophenylhydrazine. Nuclear magnetic resonance (NMR) spectra were recorded on a Varian VNMRS (300 or 500 MHz) instrument in CDCl3 or DMSO-d6. Chemical shift data were represented in units of δ (ppm) downfield from the internal standard: tetramethyl silane (TMS). Coupling constants (J) are reported in Hertz and refer to apparent peak multiplicities. Infrared (IR) spectra were measured on KBr pellets with a Varian FT-IR 660 Spectrophotometer.
Knoevenagel condensations
Following the procedure previously reported by our group (Hiller et al. 2018), it was prepared different tyrphostins by Knoevenagel condensations (Bhattacharya et al. 2014; Cabello et al. 1984; de Resende Filho et al. 2017; Deb and Bhuyan 2005; Li et al. 2011; Mukhopadhyay and Datta 2008; Ren and Cai 2007; Wang et al. 2001). The active methylene containing compound (2 mmol) (malononitrile, ethyl cyanoacetate or cyanoacetic acid), aldehyde (2 mmol) and distilled water (10 mL) were added to a rounded-bottom flask to which a reflux condenser was coupled. With the aid of an oil bath, the reaction mixture was kept at 80 °C while being magnetically stirred for 2.5–4.5 h. The progress of the reaction was monitored by Thin Layer Chromatography (TLC) using 2,4-dinitrophenylhydrazine to monitor the aldehyde consumption. After the reaction was completed, the reaction mixture was allowed to reach ambient temperature, and the product was obtained after vacuum filtration and washing with cold water. Products were dried under air. Pure samples were obtained after recrystallization with the suitable solvents. Data from the IR and NMR analyzes were compared with data from the literature, when available, as well as data obtained in our previous work (Hiller et al. 2018).
Biological assays
In vitro experiments
Mice peritoneal macrophages
Male Swiss Webster mice suffered peritoneal cavity lavage for harvesting peritoneal macrophages. This protocol adhered to the Ethical Principles in Animal Experimentation adopted by the Brazilian College of Animal Experimentation and approved by the FIOCRUZ Research Ethics Committee (number LW-033/12). The experimental protocol used was similar to Faria and collaborates in 2018 (Faria et al. 2018).
HEK 293 cells expressing human P2X7R
HEK293 cells expressing human P2X7R was maintained in according to describe for Faria and collaborates (Faria et al. 2018).
Dye uptake assay
P2X7R inhibitors and NO series compounds were pre-incubated for 10 min. P2X7R inhibitors doses ranged from 1 ng/mL to 500 μg/mL and, NO series compounds doses fluctuated from 0.01 ng/mL to 10 μg/mL. Then, mouse peritoneal macrophages (5.0 × 105 cells) were treated with 5 mM ATP for 25 min at 37 °C and propidium iodide (PI) (0.05 mg/mL in phosphate-buffered saline - PBS) or ethidium bromide (EB) incubation (25 μM in PBS), in the last 5 min of ATP treatment. HEK293 cells expressing human P2X7R (2.5 × 106 cells/mL) followed the same protocol above. These cells incubated for 24 h suffer antagonists and ATP treatment, as described for peritoneal macrophages, with posterior in the last 5 min. EB fluorescence measured with a Gemini fluorescence plate reader at an excitation wavelength of 530 nm and, an emission wavelength of 620 nm. PI fluorescence measured with a FACS Calibur Flow cytometer at an excitation wavelength of 488 nm and, an emission wavelength of 60 nm.
IL-1b enzyme-linked immunosorbent assay (ELISA)
THP-1 (human monocytic cell line derived from an acute monocytic leukemia patient) cells differentiation, activation and, maintaining were realized in according to Faria and collaborate in 2018 (Faria et al. 2018). Brilliant Blue G (BBG), A740003 (N-(1-(((Cyanoamino)(5-quinolinylamino)methylene)amino)-2,2-dimethylpropyl)-3,4-dimethoxybenzeneacetamide) and NO series compounds were added after 200 min of lipopolysaccharide (LPS) incubation, and 10 min before 5 mM ATP addition until complete 240 min of LPS incubation. The treated samples were collected, centrifuged at 1500 RPM for 5 min at 4 °C and, the supernatants were stored at −70 °C. Measuring IL-1β release using the Human IL-1 beta ELISA Kit (ab46052 -ABCAM, Cambridge). Primed peritoneal macrophages activation and treatment was similar to THP-1 cells and the mature IL-1β released quantified by sandwich ELISA following the manufacturer’s protocol (eBioscience, San Diego, CA, USA).
Caco-2 cells culture and treatments
Caco-2 cell cultured was similar to describe at Faria and collaborates in 2018 (Faria et al. 2018). Boronic acids derivatives NO-01 and NO-12, vinblastine and propranolol at 100 μM were prepared in the transport buffer (HBSS and 25 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; ELISA, enzyme-linked immunosorbent assay)) at pH 7.4 or 6.5 with 0.5% (v/v) DMSO. After Caco-2 cells incubation for 30 min, 0.3 mL of the transport buffer in the apical wells were removed and the drugs added for 60 min. LY concentrations contained in the donor and acceptor wells were measured on the plate reader M5 (molecular probes) at an excitation wavelength of 485 nm and the emission wavelength of 530 nm.
pH dependent-solubility of NO-01 and NO-12
NO-01 and NO-12 solubility was assessed from 1 to 250 μM by using DMSO (5 μL) into 995 μL buffer (pH 2.0-hydrochloride, 4.0–100 mM citrate buffer and 7.4–100 mM phosphate buffer). This solution was dispensed in a 96-well plate at room temperature for 2 h. Calibration standards were prepared by adding 5 μL of DMSO into 995 μL acetonitrile/buffer (1:1) mixture. After centrifugation (10,000 rpm, 10 min, 25 °C), the reaction samples were diluted 1:1 with acetonitrile.
Distribution coefficient (LogD) in octanol/PBS pH 7.4
A solution containing Octanol and PBS pH 7.4 at a ratio of 1:1 (v/v) was shaken mechanically for 24 h to reach the pre-saturation. Arylboronic acids analogs NO-01 and NO-12 (25 mM) in a volume of 4 μL added to 396 μL PBS for partitioning with octanol (100–400 μl). The samples were shacked; centrifuged (3000 rpm for 5 min), and after 1 h of standing the PBS layer was collected. Acetonitrile (100 μl) was added to a 100 μl aliquot of the PBS layer, and the absorbance sample was measured. The compounds were evaluated for two standard to validate the assay.
In vitro stability assays in liver microsomes
Male mice and humans liver microsomes (0.5 mg/ml in 0.1 M phosphate buffer at pH 7.4), arylboronic acids analogs NO-01 and NO-12 (1 μM) and DMSO (0.5 μM) were pre-incubated at 37 °C before NADPH addition (1 mM) to initiate the reaction in a final incubation volume of 50 μl. A buffer containing 0.1 M phosphate at pH 7.4 was used as a negative control and diazepam and verapamil as a positive control for mice and humans, respectively. Drugs were incubated for 0, 5, 15, 30, and 45 min with the microsomes and the negative control (minus NADPH) for 45 min. Methanol (50 μL) stopped the reactions at the appropriate time points and the samples centrifuged (1640×g for 20 min at 4 °C) to precipitate the proteins. Intrinsic clearance (CLint mic) for NO-01 and NO-12 microsomes was calculated according to Biosystem instructions (Faria et al. 2018).
Electrophysiological measurements
For whole-cell configuration assay was used a pipette with series resistance set as 5–11 MΏ for all the experiments in standard saline for bath and pipette solutions. Ionic currents were not compensated for recordings less than 1600 pA, however, they were compensated by 91% for values above to this value. Macrophage cell capacitance was 19.4 ± 6.24 pF; n = 73 and the holding potential of −60 mV at 37 °C for all recordings.
Saline solutions for electrophysiology
The bath solution (in mM) consisted of the following: 150 NaCl, 5 KCl, 1 MgCl2, 1 CaCl2 and, 10 HEPES (pH, 7.4) and the pipette solution (in mM): 150 KCl, 5 NaCl, 1 MgCl2, 10 HEPES and, 0.1 ethylene glycol tetraacetic acid (EGTA) pH, 7.4.
Drug application
Perfusion chamber (RC-24 chamber, Warner Instrument Corp.) was used at a rate of 1 mL/min. All the drugs were dissolved immediately before use. Compounds NO and P2X7R inhibitors were added 1–5 min before 1–5 mM ATP stimulation for 5 min at 37 °C.
In vivo experiments
Paw edema
Compounds NO were administered intraperitoneally at 60 min before intrathecal ATP (2.5 mM/ paw) administration. Mouse paw edema was measured after 30 min of ATP treatment using a plethysmometer (Insight, Brazil). This protocol adhered to the Ethical Principles in Animal Experimentation adopted by the Brazilian College of Animal Experimentation and approved by the FIOCRUZ Research Ethics Committee (number LW-58/14).
Statistical analyses
The results are presented as means ± standard deviations of the means (S.D.M.). D’Agostino and Pearson normality tests were used to test whether data followed a Gaussian distribution. In data following a Gaussian distribution, we applied an analysis of variance (ANOVA) followed by Tukey’s test. Otherwise, we applied the non-parametric Kruskal–Wallis test followed by Dunn’s test. PRISM® software (GraphPad, Inc., San Diego, CA, USA) was used for all statistical analyses.
In silico analysis
ADMET properties
The ADMET properties analysis of NO-01 and NO-12 were performed using the admetSAR (Structure-activity relationship) program, which computes a pharmacokinetic and toxicological profile (Cheng et al. 2012).
Comparative modeling
The human P2X7R model was prepared using the approach described previously (Faria et al. 2018).
Ligands molecular modeling
Chemicalize® software (ChemAxon) (Swain 2012) was used to determine the non-ionized states of the NO compounds at physiological pH 7.4. The Spartan’10 v.1.0.1 program was used to build the molecular structures. The MMFF94 (Merck Molecular Force Field) was used to generate the conformer distribution of the compounds (Halgren 1996). Then the geometry optimization was applied with the semi-empirical RM1 (Recife Model 1) method (Rocha et al. 2006).
Molecular docking
The molecular docking was performed using the algorithm MolDock (Thomsen and Christensen 2006) with the Molegro Virtual Docker (MVD) 6.0 program (CLC Bio, 8200, Aarhus, Denmark). The MolDock score [GRID] algorithm was used as the score function with a grid resolution of 0.30 Å. The partial charges were assigned according to the MVD charges scheme. The search algorithm used was the MolDock Optimizer with a search space of 40 Å around the area where the cavities of the P2X7R were found. The ligand evaluation was considered concerning internal electrostatic interactions (ES), internal hydrogen bond (HBond) and Sp2-Sp2 torsions. Every molecular docking was performed 50 runs with the same parameters set (population size = 50, max iterations = 2000, scaling factor = 0.50 and crossover rate = 0.90), followed by energy minimization. The poses binding modes were selected based on the best rank score.
Results and discussion
Synthesis
Benzene vinylnitriles were prepared by Knoevenagel condensations between aromatic aldehydes and malononitrile or ethyl cyanoacetate without catalysis and employing water as solvent. Products were obtained in high yields (> 80%) and high purity by simple vacuum filtration after reaction has reached the ambient temperature, as previously reported by our group. For the biological purposes, products were purified by recrystallization using water/ethanol or water/acetone as solvents. For the cases where two diastereoisomers (E or Z) can be formed by the Knoevenagel reactions (NO-12 and NO-13), evidence verified by NMR spectra analysis that only one stereoisomer was formed, for which E-configuration was attributed by comparison with the previously obtained datum, which had been confirmed by monocrystal X-ray diffraction.
Biological assay
In vitro
Mouse peritoneal macrophages treated with NO series compounds continuously for 72 h (Fig. 3) in the concentration of 10 μM. These analogs did not cause LDH release in this concentration, when compared with positive control for LDH release, the Triton X-100 detergent. This result indicates a low toxicity for these analogs in mammalian cells, therefore all analogs in sequence were tested in assays to evaluate the capacity of inhibiting the P2X7R function. This reduced toxicity in primary cell was consistent with previous publication. Mice peritoneal macrophages treated with the NO series compounds did not affect the cellular metabolism activity after 72 h of treatment (Hiller et al. 2018).

Cytotoxicity in mammalian cells. (A) Mice peritoneal macrophages were treated with 100 μM of the NO derivatives (NO-01, NO-03, NO-04, NO-05, NO-07, NO-11, NO-12, and NO-13 for 72 h. The graphic results represent 3–5 independent, lactate dehydrogenase (LDH) release assays. These results are expressed as mean ± s.d. ***significantly different from the negative control value at P < 0.05. CP – positive control (cells treated with 0.1% Triton X-100); CN – negative control (no treated cells)
NO series compounds caused inhibitory action on mice P2X7R activity as observed for dye uptake assay (Table 1). In addition, NO series compounds activities were compared to BBG and A740003 (selective), both P2X7R inhibitors. Therefore, the compounds NO-03, NO-05, NO-06, NO-07, NO-11 and NO-13 exhibited IC50 values superior to BBG and A740003. The compounds NO-04 and NO-08 showed IC50 values between BBG and A740003. The compounds NO-01 and NO-12 demonstrated higher inhibitory efficiency to than other compounds NO, BBG and A740003 (Table 1). ATP-induced P2X7R dye uptake was inhibited for compounds NO-01 and NO-12 with IC50 = 0.639 uM and 0.839 uM, respectively. The IC50 values for these compounds were similar to other molecules inhibitors of the P2X7R described in the literature (North 2002; Rech et al. 2016). The compounds NO-01 and NO-12 also inhibited BzATP-induced dye uptake (data not shown) with similar IC50 values observed for ATP-induced dye uptake. Therefore, NO-03, NO-05, NO-06, NO-07, NO-11, NO-13, NO-04 and NO-08 were discarded because of the reduced potency when compared to commercial P2X7R inhibitors. Contrarily, compounds NO-01 and NO-12 were tested in all complemental assays.
Dose-response curve measuring the compounds NO-01 and NO-12 toxic activity in mice peritoneal macrophages after 72 h of treatment. NO-01 and NO-12 caused low toxicity with high CC50 value (Fig. 3, Table 2). Both compounds were less toxic than BGG and A740003 when treated in the same time of incubation. When selectivity index of these both arylboronic acids derivatives were compared with commercial P2X7R inhibitors, these analogs exhibited values higher than the P2X7R inhibitors (Table 2). NO-12 analog exhibited a S.I. similar to a selective P2X7R inhibitor, A740003. NO-01 analog showed a S.I. value 1.77 times higher than compound A740003 indicating an elevate promisor activity. Therefore, these arylboronic acid analogs were tested in the subsequent assays. Curiously, both compounds showed IC50 values higher than P2X7R inhibitors previously described (Faria et al. 2018; Pacheco et al. 2018; Kwak et al. 2018; O'Brien-Brown et al. 2017), however in function of elevated S.I. value for human P2X7R, these arylboronic analogs remain as good candidates.
ATP-induced IL-1β release is a characteristic function associated with P2X7R activation. Compound NO-01 potently inhibited ATP-induced IL-1β release in mice and human P2X7R, however NO-12 weakly inhibited IL-1β release mediated by mP2X7R activation. Human THP-1 monocytes differentiated with INF-γ and PMA were primed with LPS for 4 h, with ATP added in the last 30 min of LPS incubation. Both arylboronic acids derivatives were added 30 min before ATP addition inhibited ATP-induced IL-1β release in a similar manner (Table 3). NO-1 and NO-12 exhibited higher potency than BBG and A740003 to inhibit IL-1β release mediated by hP2X7R activation (Fig. 4a). ATP-induced IL-1β release mediated by mouse P2X7R also was inhibited by arylboronic acid analogs (Fig. 4b and Table 3). NO-12 suffered a potency reduction to inhibit the mP2X7R when compared with hP2X7R. NO-1 analog maintained an elevate potency to inhibit mP2X7R with IC50 value higher than A740003 (Fig. 4, Table 3). Both analogs also inhibited BzATP-induced IL-1β release (data not shown). These results indicate a selective inhibition mechanism inhibiting IL-1β release mediated by P2X7R activation, as observed for other P2X7R antagonists (Rech et al. 2016; Volonté et al. 2012; Young and Górecki 2018).

ATP-induced IL-1β release inhibition by arylboronic acids in human and mouse cells. (a) Differentiated THP-1 cells treated with 1 mM ATP (30 min) and LPS (4 h) in the presence of increasing concentrations of BBG, A740003, NO-01, or NO-12. (b) Mouse peritoneal macrophages treated with 1 mM ATP (30 min) and LPS (4 h) in the presence of increasing concentrations of BBG, A740003, NO-01, and NO-12. Curves are representative of 3–5 independent experiments
In experiments using HEK 293 cells transfected with the human P2X7R there was potent inhibitory activity of the arylboronic acids to reduce ATP-induced dye uptake with IC50 values of 0.031 μM (NO-01) and 0.15 μM (NO-12) (Table 3). The compounds NO-01 and NO-12 conserved a high potency for inhibiting human P2X7R when compared with mouse P2X7R. A higher selectivity for human receptor has been observed for commercial or new inhibitors of the P2X7R (Gonzaga et al., 2017; Faria et al. 2018; Allsopp et al. 2017; Caseleya et al. 2016; Humphreys et al. 1998), although some commercial P2X7R antagonist may be more selective to rodents (Jiang et al. 2000; Donnelly-Roberts et al. 2009).
A possible mechanism of action for arylboronic acid analogs inhibition was measured using whole cell patch clamp technique. ATP concentrations ranging from 100 μM to 25 mM induced macroscopic ionic currents in a dose-response manner (Supplemental Fig. 1 and 2).
When arylboronic derivatives or A740003 were fixed at 500 nM and tested in the presence of ATP curve, NO-01 and NO-12 augmented the EC50 value for the ATP dose-response curve in comparison with ATP concentrations alone (Fig. 5a, b).

Evaluation of the inhibitory mechanism of compounds NO-01 and NO-12 on P2X7R antagonists. Competitive mechanisms were evaluated by recording the ionic currents of mice peritoneal macrophages with (●) ATP alone or (■) with 500 ng/mL of NO-01(A) or NO-12 (B) at 37 °C. Graphics are representative of 3–4 independent experiments
NO-01 and NO-12 compared to A740003, which is a P2X7R allosteric inhibitor acting on the pore allosteric site (Karasawa and Kawate 2016) showed action profiles similar (Figs. 5a, 4b). The ATP concentrations ranging from 10−4 to 10–1.6 were inhibited for NO-01 treatment (Supplemental Table 1), and NO-12 impaired ionic currents induced for ATP concentrations ranging from 10−3 to 10–1.7 (Supplemental Table 2).
This result indicates a P2X7R non-competitive inhibition mechanism for NO-01 and NO-12 in the allosteric site as well as the inhibitor A740003, or an irreversible interaction in the ATP binding site. This inhibitory mechanism is shared for other P2X7R antagonists recently discovered (Gonzaga et al. 2017; Faria et al. 2018; Pacheco et al. 2018).
In silico analysis
ADMET studies
The in silico pharmacokinetics evaluation indicated that the two most active compounds (NO-01 and NO-12) have a high permeability by the Human Intestinal Absorption test (Shen et al. 2010) with more than 70% being absorbed and a high permeability by the Caco-2 test (Pham The et al. 2011) with more than 8 × 10−6 cm/s. Also, high water solubility was estimated for both compounds in which the values of LogS -2.04 and − 2.20 are suggested for compounds NO-1 and NO-12, respectively. On the other hand, only compound NO-01 showed a high permeability through the blood brain barrier.
The CYP enzyme metabolism profile was also investigated in which compounds NO-1 and NO-12 only presented potential interaction with the CYP3A4 as an inhibitor. In addition, it is known that the nitrile group in most drugs is not directly metabolized, because the nitrile group is eliminated through the body unchanged. Despite that, the epoxidation of alkenenitriles compounds (such the case of compounds NO-01 and NO-12) can potentially liberate cyanide, although a large number of approved drug with alkenenitriles groups indicate that the metabolism at other sites is more likely (Fleming et al. 2010).
Regarding the toxicological parameter both compounds were labeled as weak inhibitor (pIC50 ≤ 6.0 mol/L) of the Human Ether-a-go-go-Related Gene (hERG) Inhibition Test (Marchese Robinson et al. 2011), also both compounds showed low mutagenic potential by the AMES Test and finally none carcinogenic features were detected by Carcinogens Test (Lagunin et al. 2009).
Furthermore, no apparent toxicity is identified in the literature for the boronic acids moiety regarding medical applications. Indeed, the first boronic acid-based FDA approved drug, the Bortezomib (Velcade), a proteasome inhibitor for the treatment of multiple myeloma, presented low toxicity issues. Additionally, the eventual final product in the metabolism of boronic acid-containing compounds is generally boric acid, which has low particularly toxic to humans (Cambre and Sumerlin 2011).
Solubility, microsomal stability, and permeability in vitro
Arylboronic acids NO-01 and NO-12 were moderately stable in mouse and human microsomes (Table 4). NO-01 when compared to NO-12 may exhibit a short duration of action in vivo. Additionally, NO-01 was permeable in Caco-2 at percentages above to 75% (Table 4) and NO-12 was permeable in a range superior to 55%, both when compared with propranolol.
When solubilized in solutions with pH values ranging from 2 to 10, NO-01 demonstrated solubility in turn of 250 μM (Table 5). LogD7.4 measured for NO-01 gave a value of −2.25 ± 0.09 indicating good solubility in aqueous solutions (Table 6). In relation to NO-12, it demonstrated solubility at concentrations above 250 μM (Table 5). The LogD7.4 value for NO-12 was −0.96 ± 0.11 (Table 6).
NO-01 reduced lipophilicity may be associated with short microsomal stability, however this characteristic can increase the tissue permeability as observed in Caco-2 cells. In contrast, NO-12 although lipophilic exhibited higher microsomal stability and reduced tissue permeability.
In vivo assays
NO-01 and NO-12 anti-inflammatory properties in vivo was evaluated using the ATP- induced paw edema model in mice. Sodium diclofenac and arylboronic acids were administered intraperitoneally at 1 h before ATP treatment for 30 min. The oxidized ATP, an irreversible P2X7R antagonist inhibited the ATP-induced inflammatory response (NO-01, Fig. 6a and NO-12, Fig. 6b). Sodium diclofenac, a general anti-inflammatory, also inhibited ATP-induced paw edema (Fig. 6). NO-01 and NO-12 dose-dependently inhibited the ATP effect (Fig. 6b). In this context, NO-01 promoted an anti-inflammatory effect more potent than oxidized ATP and diclofenac (Fig. 6a). In accordance with in vitro assays, NO-01 inhibited the ATP-induced paw edema with higher potency compared to the NO-12 analog. Triazole and naphtoquinone derivatives inhibiting P2X7R activity also reduced the ATP-induced paw edema (Gonzaga et al., 2017, Faria et al. 2018). In both papers, at least one dose administered reducing edema formation in μg/kg concentrations as observed mainly for NO-01. These arylboronic acid analogs also inhibited carrageenan-induced response in similar manner to ATP stimulation (data not shown).

Anti-inflammatory effects of NO-01 and NO-12 on ATP-induced paw edema in mice. Groups with five Swiss Webster mice were treated with ATP (intraplantar) or preincubated for 30 min with Diclofenac (10 mg/kg), oxidized ATP (10 mg/kg), or increasing doses of NO-01 (0.001–1 mg/kg) (a) or NO-12 (b). Paw edema was measured at 60 min after ATP application. These results represent three distinct days and are expressed as mean ± s.d. ### P < 0.05 comparison in relation to saline group. *** P < 0.05 comparison in relation to ATP group
Molecular docking
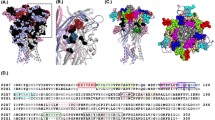
The blind molecular docking of the two most active ligands (NO-01 and NO-12), on the ionic channel P2X7R, were performed around the area where the cavities were identified in the protein molecular structure (Fig. 7). In order to harness structure activity relationship the same blind molecular docking were performed with less active ligands (NO-06 and NO-13). The largest volume cavity is the S1 located into the P2X7 pore comprised by the lower body domain of the three correlated subunits; the second largest volume cavity is the S2, also located in the pore, but in the upper body domain and; the cavities S3´, S3´´ and S3´´´ are the ATP-binding sites located between the protein subunits. Figure 7 depicts the cavities identified in the P2X7R and the Table 7 indicates the volume of each cavity.

Representation of the binding sites (S1, S2 and S3) identified in the P2X7R. Frontal view (a) and Top view (b). The cavities S1, S2 and S3 are ordered based on their volumes. Because there are three identical S3 binding sites formed by three identical subunits, they are labeled S3`, S3``, S3```
The molecular docking results indicated that the cavity S2 was the most favorable binding site of the inhibitors NO-01 NO-12, NO-06 and NO-13. The S2 cavity is the same cavity revealed in the work from Karasawa and Kawate (2016) where the five known P2X7 inhibitors bind (A740003, A804598, AZ10606120, GW791343, JNJ47965567), based on the elucidation of the panda P2X7R complexed crystal structures.
All the four NO compounds presented a similar binding mode into the S2 cavity, with their phenyl moiety interacting with the phenylalanine residues 95, 103 and 293 from chain B. Also the boronic acid group from the inhibitors NO-01, NO-12 and NO-13 are orientated towards the serine 101 lateral chain from chain B (Fig. 8).

Superposition of the inhibitors NO-01, NO-12, NO-06 and NO-13 docked into the S2 cavity. In blue are depicted the NO derivatives and in green the main residues that are in close contact
Some residues interactions with the NO compounds are similar to the known allosteric inhibitors identified from the Karasawa and Kawate (2016) work, as the phenylalanines 95, 103 and 293 are also involved in hydrophobic interactions. In addition, the residue Met105 that performs hydrophobic interactions with the compounds A740003, A804598 and JNJ47965567 is also interacting with the compound NO-06.
Despite the fact that all four NO compounds submitted to molecular docking studies presented similar hydrophobic interactions with the phenylalanine residues, the hydrogen bond formation with the residues Gln98, Leu97 and specially with the Ser101 might be responsible for the gain in the activity of the compounds NO-01 and NO-12 compared with the compounds NO-06 and NO-13. Also, the inhibitor NO-12 presented extra hydrogen bonds with the residues glycine 98 and 99 from chain A, which might contribute for selectivity. Figure 9 depicts the intermolecular interaction of the NO compounds into de S2 binding cavity found by molecular docking.

Representation of the main intermolecular interactions of each NO compounds into de S2 binding cavity, identified from the molecular docking. The arrows indicates hydrogen bonds and the blue areas in the NO structures indicates solvent exposure
Though the molecular docking results indicate the probable binding site of NO series compounds in the human P2X7R model and suggest that interactions of the boronic acid moiety with Ser101 and Gln98 may be determinant to high affinity of the compounds NO-01 and NO-12, other studies in future should be done to refine these insights in order to increase the information on the properties and inhibitory activity of the boronic acids derivatives on the P2X7R.
Conclusion
In summary, the results exhibited herein demonstrate arylboronic acids as a novel P2X7R inhibitors class. NO-01 and NO-12 compounds ATP antagonistic effects resulted in potent blockage of mouse and human P2X7R. The arylboronic acid NO-01 was more potent in vitro and in vivo when compared with BBG and A740003 to reduce P2X7R activity and the inflammatory response. In according to in silico ADMET analysis both molecules depicted an acceptable pharmacokinetic profile and low toxicological effects, thus suggesting its potential drug-likeness characteristic. The molecular docking results suggest that the binding sites for the compounds NO-01 and NO-12 are located in the upper area of P2X7R pore. Thus, these results strengthening the therapeutic potential and the feasibility of developing new P2X7R inhibitors based on the arylboronic acid pharmacophore group.
References
Ahmed V, Liu Y, Silvestro C, Taylor SD (2006) Boronic acids as inhibitors of steroid sulfatase. Bioorg Med Chem 14:8564–8573. https://doi.org/10.1016/j.bmc.2006.08.033
Allsopp RC, Dayl S, Schmid R, Evans RJ (2017) Unique residues in the ATP gated human P2X7 receptor define a novel allosteric binding pocket for the selective antagonist AZ10606120. Sci Rep 7:725
Asano T, Nakamura H, Uehara Y, Yamamoto Y (2004) Design, synthesis, and biological evaluation of aminoboronic acids as growth-factor receptor inhibitors of EGFR and VEGFR-1 tyrosine kinases. Chembiochem Eur J Chem Biol 5:483–490. https://doi.org/10.1002/cbic.200300748
Baas T (2012) Paradoxical P2X7. Sci-Bus Exch 5:512–512. https://doi.org/10.1038/scibx.2012.512
Baker SJ, Ding CZ, Akama T, Zhang Y-K, Hernandez V, Xia Y (2009) Therapeutic potential of boron-containing compounds. Future Med Chem 1:1275–1288. https://doi.org/10.4155/fmc.09.71
Ban HS, Nakamura H (2015) Boron-based drug design. Chem Rec N Y N 15:616–635. https://doi.org/10.1002/tcr.201402100
Ban HS, Usui T, Nabeyama W, Morita H, Fukuzawa K, Nakamura H (2009) Discovery of boron-conjugated 4-anilinoquinazoline as a prolonged inhibitor of EGFR tyrosine kinase. Org Biomol Chem 7:4415–4427. https://doi.org/10.1039/b909504g
Bartlett R, Stokes L, Sluyter R (2014) The P2X7 receptor channel: recent developments and the use of P2X7 antagonists in models of disease. Pharmacol Rev 66:638–675. https://doi.org/10.1124/pr.113.008003
Baudelet D, Lipka E, Millet R, Ghinet A (2015) Involvement of the P2X7 purinergic receptor in inflammation: an update of antagonists series since 2009 and their promising therapeutic potential. Curr Med Chem 22:713–729
Bhattacharya B, Maity DK, Pachfule P, Colacio E, Ghoshal D (2014) Syntheses, X-ray structures, catalytic activity and magnetic properties of two new coordination polymers of Co(II) and Ni(II) based on benzenedicarboxylate and linear N,N′-donor Schiff base linkers. Inorg Chem Front 1:414–425. https://doi.org/10.1039/c4qi00032c
Bradke TM, Hall C, Carper SW, Plopper GE (2008) Phenylboronic acid selectively inhibits human prostate and breast cancer cell migration and decreases viability. Cell Adhes Migr 2:153–160
Buac D, Shen M, Schmitt S, Kona FR, Deshmukh R, Zhang Z, Neslund-Dudas C, Mitra B, Dou QP (2013) From bortezomib to other inhibitors of the proteasome and beyond. Curr Pharm Des 19:4025–4038
Burnstock G, Knight GE (2018) The potential of P2X7 receptors as a therapeutic target, including inflammation and tumour progression. Purinergic Signal 14:1–18. https://doi.org/10.1007/s11302-017-9593-0
Cabello JA, Campelo JM, Garcia A, Luna D, Marinas JM (1984) Knoevenagel condensation in the heterogeneous phase using aluminum phosphate-aluminum oxide as a new catalyst. J Org Chem 49:5195–5197. https://doi.org/10.1021/jo00200a036
Cambre JN, Sumerlin BS (2011) Biomedical applications of boronic acid polymers. Polymer 52:4631–4643. https://doi.org/10.1016/j.polymer.2011.07.057
Caseleya EA, Muencha SP, Fishwickb CW, Jiang L (2016) Structure-based identification and characterisation of structurally novel human P2X7 receptor antagonists. Biochem Pharmacol 116(15):130–139
Chen Z, He L, Li L, Chen L (2018) The P2X7 purinergic receptor: an emerging therapeutic target in cardiovascular diseases. Clin Chim Acta Int J Clin Chem 479:196–207. https://doi.org/10.1016/j.cca.2018.01.032
Cheng F, Li W, Zhou Y, Shen J, Wu Z, Liu G, Lee PW, Tang Y (2012) admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model 52:3099–3105. https://doi.org/10.1021/ci300367a
Das BC, Thapa P, Karki R, Schinke C, Das S, Kambhampati S, Banerjee SK, Van Veldhuizen P, Verma A, Weiss LM, Evans T (2013) Boron chemicals in diagnosis and therapeutics. Future Med Chem 5:653–676. https://doi.org/10.4155/fmc.13.38
de Resende Filho JBM, Pires GP, de Oliveira Ferreira JMG, Teotonio EES, Vale JA (2017) Knoevenagel condensation of aldehydes and ketones with Malononitrile catalyzed by amine compounds-tethered Fe3O4@SiO2 nanoparticles. Catal Lett 147:167–180. https://doi.org/10.1007/s10562-016-1916-1
Deb, M.L., Bhuyan, P.J., 2005. Uncatalysed Knoevenagel condensation in aqueous medium at room temperature - PDF free download [WWW document]. Tetrahedron Lett. URL https://www.sciencedirect.com/science/article/pii/S004040390501631X (accessed 9.20.18)
Diaz DB, Yudin AK (2017) The versatility of boron in biological target engagement. Nat Chem 9:731–742. https://doi.org/10.1038/nchem.2814
Dimitrova P, Ivanovska N (2013) Tyrphostins as a promising therapeutic tool in inflammation-related conditions. OA Inflamm 1. https://doi.org/10.13172/2052-787X-1-1-608
Donnelly-Roberts DL, Namovic MT, Surber B, Vaidyanathan SX, Perez-Medrano A, Wang Y, Carroll WA, Jarvis MF (2009) [3H]A-804598 ([3H]2-cyano-1-[(1S)-1-phenylethyl]-3-quinolin-5-ylguanidine) is a novel, potent, and selective antagonist radioligand for P2X7 receptors. Neuropharm. 56:223–229
Dou QP, Zonder JA (2014) Overview of proteasome inhibitor-based anti-cancer therapies: perspective on bortezomib and second generation proteasome inhibitors versus future generation inhibitors of ubiquitin-proteasome system. Curr. Cancer Drug Targets 14:517–536
Faria RX, Freitas HR, Reis RAM (2017) P2X7 receptor large pore signaling in avian Müller glial cells. J Bioenerg Biomembr 49:215–229. https://doi.org/10.1007/s10863-017-9717-9
Faria RX, Oliveira FH, Salles JP, Oliveira AS, von Ranke NL, Bello ML, Rodrigues CR, Castro HC, Louvis AR, Martins DL, Ferreira VF (2018) 1,4-naphthoquinones potently inhibiting P2X7 receptor activity. Eur J Med Chem 143:1361–1372. https://doi.org/10.1016/j.ejmech.2017.10.033
Fleming FF, Yao L, Ravikumar PC, Funk L, Shook BC (2010) Nitrile-containing pharmaceuticals: efficacious roles of the nitrile pharmacophore. J Med Chem 53:7902–7917. https://doi.org/10.1021/jm100762r
Gonzaga DTG, Gomes LB, Costa TEMC, von Ranke NL, Pacheco PAF, Simões APS, Arruda JC, Dantas LP, de Freiras HR, Reis RA, Penido C, Bello ML, Castro HC, Rodrigues CRR, Ferreira VF, Faria RX, Silva FC (2017) 1-Aryl-1H- and 2-aryl-2H-1,2,3-triazole derivatives blockade P2X7 receptor in vitro and inflammatory response in vivo. Eur J Med Chem 139:698–717. https://doi.org/10.1016/j.ejmech.2017.08.034
Gorodeski GI (2012) P2X7 receptors and epithelial cancers. Wiley Interdiscip Rev Membr Transp Signal 1:349–371. https://doi.org/10.1002/wmts.33
Groziak MP (2001) Boron therapeutics on the horizon. Am J Ther 8:321–328
Gyurkovska V, Stefanova T, Dimitrova P, Danova S, Tropcheva R, Ivanovska N (2014) Tyrosine kinase inhibitor tyrphostin AG490 retards chronic joint inflammation in mice. Inflammation 37:995–1005. https://doi.org/10.1007/s10753-014-9820-6
Halgren TA (1996) Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J Comput Chem 17:490–519. https://doi.org/10.1002/(SICI)1096-987X(199604)17:5/6<490::AID-JCC1>3.0.CO;2-P
Hiller N d J, Silva NAAE, Faria RX, Souza ALA, Resende JALC, Borges Farias A, Correia Romeiro N, de Luna Martins D (2018) Synthesis and evaluation of the anticancer and Trypanocidal activities of Boronic Tyrphostins. ChemMedChem 13:1395–1404. https://doi.org/10.1002/cmdc.201800206
Humphreys, B.D., Virginio, C., Surprenant, A., Rice, J., Dubyak, G.R., 1998. Isoquinolines as antagonists of the P2X7 nucleotide receptor: high selectivity for the human versus rat receptor homologues. Mol Pharmacol. Jul;54(1):22-32
Jiang LH, MacKenzie AB, North RA, Surprenant A (2000) BBG selectively blocks ATP-gated rat P2X7 receptors. Mol Pharmacol 58:82–88
Karasawa A, Kawate T (2016) Structural basis for subtype-specific inhibition of the P2X7 receptor. eLife 5. https://doi.org/10.7554/eLife.22153
Kim M, Jiang LH, Wilson HL, North RA, Surprenant A (2001) Proteomic and functional evidence for a P2X7 receptor signalling complex. EMBO J 20:6347–6358. https://doi.org/10.1093/emboj/20.22.6347
Koehler KA, Lienhard GE (1971) 2-phenylethaneboronic acid, a possible transition-state analog for chymotrypsin. Biochemistry 10:2477–2483
Kumar SK, Hager E, Pettit C, Gurulingappa H, Davidson NE, Khan SR (2003) Design, synthesis, and evaluation of novel boronic-chalcone derivatives as antitumor agents. J Med Chem 46:2813–2815. https://doi.org/10.1021/jm030213+
Kwak SH, Shin S, Lee JH, Shim JK, Kim M, Lee SD, Lee A, Bae J, Park JH, Abdelrahman A, Müller CE, Cho SK, Kang SG, Bae MA, Yang JY, Ko H, Goddard WA, Kim YC (2018) Synthesis and structure-activity relationships of quinolinone and quinoline-based P2X7 receptor antagonists and their anti-sphere formation activities in glioblastoma cells. Eur J Med Chem 151:462–481
Lagunin A, Filimonov D, Zakharov A, Xie W, Huang Y, Zhu F, Shen T, Yao J, Poroikov V (2009) Computer-aided prediction of rodent carcinogenicity by PASS and CISOC-PSCT. QSAR Comb Sci 28:806–810. https://doi.org/10.1002/qsar.200860192
LeBeau AM, Singh P, Isaacs JT, Denmeade SR (2008) Potent and selective peptidyl Boronic acid inhibitors of the serine protease prostate-specific antigen. Chem Biol 15:665–674. https://doi.org/10.1016/j.chembiol.2008.05.020
Li G, Xiao J, Zhang W (2011) Knoevenagel condensation catalyzed by a tertiary-amine functionalized polyacrylonitrile fiber. Green Chem 13:1828–1836. https://doi.org/10.1039/C0GC00877J
Marchese Robinson RL, Glen RC, Mitchell JBO (2011) Development and comparison of hERG blocker classifiers: assessment on different datasets yields markedly different results. Mol Inform 30:443–458. https://doi.org/10.1002/minf.201000159
Matthews DA, Alden RA, Birktoft JJ, Freer ST, Kraut J (1975) X-ray crystallographic study of boronic acid adducts with subtilisin BPN’ (novo). A model for the catalytic transition state J Biol Chem 250:7120–7126
Mehta N, Kaur M, Singh M, Chand S, Vyas B, Silakari P, Bahia MS, Silakari O (2014) Purinergic receptor P2X7: a novel target for anti-inflammatory therapy. Bioorg Med Chem 22:54–88. https://doi.org/10.1016/j.bmc.2013.10.054
Modzelewska A, Pettit C, Achanta G, Davidson NE, Huang P, Khan SR (2006) Anticancer activities of novel chalcone and bis-chalcone derivatives. Bioorg Med Chem 14:3491–3495. https://doi.org/10.1016/j.bmc.2006.01.003
Mukhopadhyay C, Datta A (2008) A simple, efficient and green procedure for the Knoevenagel condensation of aldehydes with N-Methylpiperazine at room temperature under solvent-free conditions. Synth Commun 38:2103–2112. https://doi.org/10.1080/00397910802029364
Nakamura H, Kuroda H, Saito H, Suzuki R, Yamori T, Maruyama K, Haga T (2006) Synthesis and biological evaluation of boronic acid containing cis-stilbenes as apoptotic tubulin polymerization inhibitors. ChemMedChem 1:729–740. https://doi.org/10.1002/cmdc.200600068
North RA (2002) Molecular physiology of P2X receptors. Physiol Rev 82:1013–1067. https://doi.org/10.1152/physrev.00015.2002
O'Brien-Brown, J., Jackson, A., Reekie, T.A., Barron, M.L., Werry, E.L., Schiavini, P., McDonnell, M., Munoz, L., Wilkinson, S., Noll, B., Wang, S., Kassiou, M., 2017. Discovery and pharmacological evaluation of a novel series of adamantyl cyanoguanidines as P2X7 receptor antagonists. Eur J med Chem. Apr 21;130:433-439
Pacheco PAF, Galvão RMS, Faria AFM, von Ranke, NL, Rangel MS, Ribeiro TM, Bello ML, Rodrigues CF, Ferreira VF, da Rocha DR, Faria RX (2018) 8-Hydroxy-2-(1H-1,2,3-triazol-1-yl)-1,4-naphtoquinone derivatives inhibited P2X7 Receptor-Induced dye uptake into murine Macrophages. Bioorg Med Chem 27:1449–1455
Pham The H, González-Álvarez I, Bermejo M, Mangas Sanjuan V, Centelles I, Garrigues TM, Cabrera-Pérez MÁ (2011) In silico prediction of Caco-2 cell permeability by a classification QSAR approach. Mol. Inform. 30:376–385. https://doi.org/10.1002/minf.201000118
Philipp M, Bender ML (1971) Inhibition of serine proteases by arylboronic acids. Proc Natl Acad Sci U S A 68:478–480
Rech JC, Bhattacharya A, Letavic MA, Savall BM (2016) The evolution of P2X7 antagonists with a focus on CNS indications. Bioorg Med Chem Lett 26:3838–3845. https://doi.org/10.1016/j.bmcl.2016.06.048
Ren Y, Cai C (2007) Knoevenagel condensation of aromatic aldehydes with active methylene compounds using a catalytic amount of iodine and K 2 CO 3 at room temperature. Synth Commun 37:2209–2213. https://doi.org/10.1080/00397910701397375
Rocha GB, Freire RO, Simas AM, Stewart JJP (2006) RM1: a reparameterization of AM1 for H, C, N, O, P, S, F, cl, Br, and I. J Comput Chem 27:1101–1111. https://doi.org/10.1002/jcc.20425
Savio LEB, de Andrade Mello P, da Silva CG, Coutinho-Silva R (2018) The P2X7 receptor in inflammatory diseases: angel or demon? Front Pharmacol 9:52. https://doi.org/10.3389/fphar.2018.00052
Shemon AN, Sluyter R, Fernando SL, Clarke AL, Dao-Ung L-P, Skarratt KK, Saunders BM, Tan KS, Gu BJ, Fuller SJ, Britton WJ, Petrou S, Wiley JS (2006) A Thr357 to Ser polymorphism in homozygous and compound heterozygous subjects causes absent or reduced P2X7 function and impairs ATP-induced mycobacterial killing by macrophages. J Biol Chem 281:2079–2086. https://doi.org/10.1074/jbc.M507816200
Shen J, Cheng F, Xu Y, Li W, Tang Y (2010) Estimation of ADME properties with substructure pattern recognition. J Chem Inf Model 50:1034–1041. https://doi.org/10.1021/ci100104j
Shimizu K, Maruyama M, Yasui Y, Minegishi H, Ban HS, Nakamura H (2010) Boron-containing phenoxyacetanilide derivatives as hypoxia-inducible factor (HIF)-1alpha inhibitors. Bioorg Med Chem Lett 20:1453–1456. https://doi.org/10.1016/j.bmcl.2009.12.037
Skaper SD (2011) Ion channels on microglia: therapeutic targets for neuroprotection. CNS Neurol. Disord. Drug Targets 10:44–56
Suenaga H, Nakashima K, Mikami M, Yamamoto H, James TD, Sandanayake KRAS, Shinkai S (1996) Screening of arylboronic acids to search for a strong inhibitor for γ-glutamyl transpeptidase (γ-GTP). Recl Trav Chim Pays-Bas-J R Neth 115:44–48
Sugiyama T (2014) Role of P2X7 receptors in the development of diabetic retinopathy. World J Diabetes 5:141–145. https://doi.org/10.4239/wjd.v5.i2.141
Swain M (2012) Chemicalize.Orgchemicalize.Orgby ChemAxon ltd. J Chem Inf Model 52:613–615. https://doi.org/10.1021/ci300046g
Thomsen R, Christensen MH (2006) MolDock: a new technique for high-accuracy molecular docking. J Med Chem 49:3315–3321. https://doi.org/10.1021/jm051197e
Tsuda M, Tozaki-Saitoh H, Inoue K (2010) Pain and purinergic signaling. Brain Res Rev 63:222–232. https://doi.org/10.1016/j.brainresrev.2009.11.003
Volonté C, Apolloni S, Skaper SD, Burnstock G (2012) P2X7 receptors: channels, pores and more. CNS Neurol. Disord. Drug Targets 11:705–721
Wang S, Ren Z, Cao W, Tong W (2001) The Knoevenagel condensation of aromatic aldehydes with Malononitrile or ethyl Cyanoacetate in the presence of Ctmab in water. Synth Commun 31:673–677. https://doi.org/10.1081/SCC-100103255
Weston GS, Blázquez J, Baquero F, Shoichet BK (1998) Structure-based enhancement of boronic acid-based inhibitors of AmpC beta-lactamase. J Med Chem 41:4577–4586. https://doi.org/10.1021/jm980343w
Young CNJ, Górecki DC (2018) P2RX7 Purinoceptor as a therapeutic target-The second coming? Front Chem 6:248. https://doi.org/10.3389/fchem.2018.00248
Zervosen A, Sauvage E, Frère J-M, Charlier P, Luxen A (2012) Development of new drugs for an old target: the penicillin binding proteins. Mol Basel Switz 17:12478–12505. https://doi.org/10.3390/molecules171112478
Acknowledgements
CAPES, CNPq, FAPERJ, UFRJ, UFF.
Funding
This work was supported by CNPq (National Council of Research of Brazil) (Fellowship Process Number 304716/2014–6).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplemental Figure 1
Representative macroscopic ionic currents obtained from peritoneal macrophages treated with ATP alone or in the presence of P2X7R antagonists. The arrow represents the moment of ATP addition. NO-01 and A740003 were added 5 minutes before ATP application. All recordings were measured in a holding potential of – 60 mV.
Supplemental Figure 2
Representative macroscopic ionic currents obtained from peritoneal macrophages treated with ATP alone or in the presence of P2X7R antagonists. The arrow represents the moment of ATP addition. NO-12 and A740003 were added 5 minutes before ATP application. All recordings were measured in a holding potential of – 60 mV.
ESM 3
(DOC 122 kb)
Rights and permissions
About this article

Cite this article
Faria, R.X., de Jesus Hiller, N., Salles, J.P. et al. Arylboronic acids inhibit P2X7 receptor function and the acute inflammatory response. J Bioenerg Biomembr 51, 277–290 (2019). https://doi.org/10.1007/s10863-019-09802-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10863-019-09802-x