
Abstract
A blood pre-centrifugation delay of 24 h at room temperature influenced the proton NMR spectroscopic profiles of human serum. A blood pre-centrifugation delay of 24 h at 4°C did not influence the spectroscopic profile as compared with 4 h delays at either room temperature or 4°C. Five or ten serum freeze–thaw cycles also influenced the proton NMR spectroscopic profiles. Certain common in vitro preanalytical variations occurring in biobanks may impact the metabolic profile of human serum.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Human biological fluid or tissue metabolomics can be successfully used in the identification of clinically relevant biomarkers by profiling spectral differences between case and control individuals (Griffin and Shockor 2004). One of the main methods used in metabolic profiling, which can be efficiently used with biological fluids and tissues, is proton NMR spectroscopy (Lindon et al. 2001; Howe and Opstad 2003; Piotto et al. 2008; Fujiwara et al. 2009). Metabolomics, through proton NMR spectroscopy, has been used efficiently in clinical and toxicological studies (Gartland et al. 1990; Holmes et al. 1998), clinical chemistry applications (Kaartinen et al. 1998), pharmacological studies (Le Moyec et al. 2005) and corresponding metabolomic data analysis procedures have been described (Lindon et al. 2005). Metabolic profiles (metabotypes) are influenced not only by disease-related processes, such as metabolic diseases, but also by the genetic background (Gavaghan et al. 2000) and by in vivo preanalytical variations including food and drug intake, exercise and stress (Walsh et al. 2006).
However, very little is known about the impact of in vitro preanalytical variations on proton NMR spectroscopy analytical endpoints (Teahan et al. 2006; Barton et al. 2008). in vitro preanalytical variations are under the direct control of biobanks. Biobanks are organized collectors and providers of biospecimens for research purposes. They collect, authenticate, preserve and offer independent access to biological materials for a variety of technological applications. An important number of samples are often required to reach meaningful and reproducible results. Therefore researchers may need samples from different collections or from different biobank settings. These samples have almost always undergone different preanalytical treatments. Most biobanks store samples to support different research purposes and do not apply specific processing methods currently used by metabolomics laboratories, such as deproteination by ultrafiltration or acetonitrile. In this respect, it is important for biobanks to have knowledge on the suitability of a sample for specific downstream applications, in this case metabolomics. It is also important for researchers to know if they can safely use samples with different preanalytical treatments without introducing a bias linked to systematic differences in sample collection and processing. The most commonly encountered and more important pre-analytical variations can be captured through a Standard PREanalytical Code (SPREC) which has recently been described (Betsou 2010). We examined the impact of common in vitro preanalytical variations, namely pre-centrifugation conditions and freeze–thaw cycles, on serum metabolic profiles using proton NMR spectroscopy.
Experimental
Biological samples
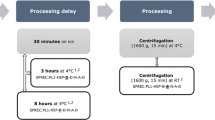
Blood from 7 healthy donors (3 male and 4 female; median age 30 [24–52]) was collected in SST tubes. All donors signed an informed consent release form and the study was approved by the ethics committee and the French Biomedicine Agency (protocol no PFS09-010). Six SST tubes were collected from each donor. Of these tubes, three stood at room temperature for 4 h (more blood was collected under these conditions because they were considered as the reference), one stood at room temperature for 24 h, one was stored at 4°C for 4 h and one at 4°C for 24 h. All storage conditions were executed before centrifugation. Centrifugation was performed at 2000×g at room temperature for 10 min. Immediately after centrifugation, serum was aliquoted in 0.2 ml aliquots and stored at −80°C in polypropylene cryotubes. The SPREC for the four different conditions were SER-SST-C-B-N-B-A, SER-SST-I-B-N-B-A, SER-SST-D-B-N-B-A and SER-SST-J-B-N-B-A respectively (Betsou 2010). Aliquots were prepared from the SER-SST-D-B-N-B-A, SER-SST-I-B-N-B-A and SER-SST-J-B-N-B-A samples. From the pooled and homogenized SER-SST-C-B-N-B-A sample, some aliquots were submitted to four freeze–thaw cycles and others were submitted to nine freeze–thaw cycles. Thawing took place at room temperature for 30 min and the samples were re-frozen at −80°C (Sukumaran et al. 2009). All samples were thawed at the moment of analysis. The same pooled and homogenized sample was used for the pre-centrifugation delay analysis. The experimental plan is shown in Table 1.
Processing of serum samples for proton NMR
One 0.2 ml aliquot of serum was thawed at ambient temperature, diluted in 0.3 ml of D2O and homogenized by vortexing for 3 min. The pH was adjusted to 7 with a 0.1 N DCl solution in order to reduce shifts and to correspond to the pH used in the database. All final volumes were adjusted with D2O. After homogenization and centrifugation at 20160×g for 5 min, the supernatant was transferred to NMR 509-UP Norell tubes (equivalent to 535-PP Wilmad tubes) purchased from Eurisotop (91194 Saint Aubin cedex, France). A capillary containing 1 mg/ml 3-(trimethylsilyl) 3,3,3,3-tetradeutero-propionic acid (TSP) in D2O, as an internal reference, was introduced in the NMR 509-UP tube. TSP was used in a capillary in order to avoid TSP interference with albumin (Kriat et al. 1992; Goldsmith et al. 2009).
Spectroscopic analysis
NMR spectra were acquired at 300 K on a Bruker AVANCE III 600 spectrometer (Magnet system 14.09 T 600 MHz/54 mm) equipped with a TXI 5 mm z-gradient probe. The TOPSPIN (Bruker) software was used. Shim control was performed automatically by gradient shimming and final lineshape optimization (Topshim 1D procedure). Samples were first analyzed with a classical proton sequence (90° proton pulse was calibrated to 7.36 μs at −1 dB (18.34 W) then with a Carr-Purcell-Meiboom-Gill (CPMG) T2-filter sequence with an attenuation of the high molecular weight/lipid molecule effects on the spectra. For each sample, a one-dimensional proton spectrum using the CPMG pulse sequence was acquired from 128 scans containing 64 k data points, a spectral width of 6,009 Hz and relaxation delay, D1, of 2 s. The inter-pulse delay between the 180° pulses of the CPMG pulse train was synchronized with the sample rotation and set to 1,400 μs. The number of loops was set to 200 so the CPMG pulse train had a total length of 283 ms. The FID was multiplied by an exponential weighing function corresponding to 0.3 Hz prior to Fourier transform. The non-zero filled obtained spectra were manually phased and baseline-corrected, calibrated to TSP at 0.00 ppm, all using XWIN NMR (version 3.3, Bruker) and finally bucketed. Bucketed spectra were separated into peaks and each peak area was calculated; each peak area corresponded to specific chemical displacements.
Furthermore, two-dimensional J-resolved NMR spectra were acquired with a 1.0 s relaxation delay using 32 scans per 128 increments that were collected into 16 K data points using spectral widths of 10,000 Hz in F2 and 78 Hz in F1. Spectra were recorded at 300 K. J-resolved spectra were tilted by 45° and symmetrized about F1. Coherence order selective gradient heteronuclear single quantum coherence (HSQC) spectra were recorded for a data matrix of 256 × 16,384 points covering 30,185 × 8,013 Hz with 92 scans for each increment. INEPT transfer delays were optimized for a heteronuclear coupling of 145 Hz and a relaxation delay of 1.5 s was applied. Data was linearly predicted in F1 to 512 × 16,384 using 32 coefficients and then zero-filled to 2,048 × 16,384 points prior to echo-anti echo type 2D Fourier transformation. A sine bell shaped window function, shifted by π/2 in both dimensions, was then applied.
NMR methods show a high reproducibility through compound reference lineshape systematic control while operator variability has been shown to be negligible compared with the intrinsic variability (Sukumaran et al. 2009).
Data analysis
The 1H-NMR spectra were automatically reduced to ASCII files using MestReNova (v. 5.2.5, Mestrelab Research, Santiago de Compostela, Spain). Spectral intensities were scaled to TSP and reduced to integrated regions or “buckets” of equal width (0.04 ppm) corresponding to the region of d 9.6 to d 0.0. The region from δ 4.85 ppm to δ 4.75 ppm was removed from the analysis due to the residual signal of water. Proton signals corresponding to TSP-d4 (at δ 0.00 ppm) were also removed. The generated ASCII file was imported into Microsoft Excel (Microsoft) for the addition of labels. Principal component analysis (PCA) and partial least squares discriminant analysis (PLS-DA) were performed with the SIMCA-P software (v. 11.0, Umetrics, Umea, Sweden) (Tenenhaus et al. 1995). The scaling method for PCA and for PLS-DA was Pareto. The PLS-DA model was validated using the permutation method through 20 applications. Each signal identified by PLS-DA as discriminant was integrated using MestReNova. The resulting area was used to test the significance of the different metabolites selected by a Mann–Whitney test using the Excel software. Identification of the different metabolites was obtained via data comparison with NMR spectra of pure reference compounds and with standard metabolite chemical shift tables of the HMDB database (Wishart et al. 2009).
Results and discussion
A representative spectrum of human serum obtained by 1H NMR analysis is shown in Fig. 1. Several metabolites were identified (Table 2; Fig. 1). Although certain metabolites could not be identified due to the superposition of spectral peaks, most of the metabolite assignments were in agreement with previously published profiles (He et al. 2011, Psychogios et al. 2011). PCA is a non-targeted analysis and does not require identification of signals; it only requires processed numerical data from spectra intensity and identifies the directions of maximum variability. We selected 3 PCs for the NMR spectra which explained 75.7% of the total variance; however the comprehensive profiles indicated that samples could not be differentiated by PCA; samples of the four different preanalytical treatments, SER-SST-C-B-N-B-A, SER-SST-D-B-N-B-A, SER-SST-I-B-N-B-A and SER-SST-J-B-N-B-A were similarly distributed. As can be seen in Fig. 2a, there was more assembly between samples from the same donor than between samples of the same preanalytical conditions. For instance, donor 1 is clearly separated from the other donors along the x-axis and the separations are even more distinguishable in the PLS-DA score plot (See supplementary data). Preanalytical variability was negligible compared with inter-individual variability. In this case, a supervised multivariate data analysis using covariance is required to try to associate a group of metabolites to specific preanalytical conditions. Partial least square discriminant analysis (PLS-DA) corresponds to such a supervised method. In contrast to PCA, which only uses information from the metabolomic matrix, PLS-DA also takes into account information from the matrix of the preanalytical condition types. PLS-DA showed a significant grouping of samples of the SER-SST-I-B-N-B-A preanalytical type. These samples corresponded to serum having stood at room temperature for 24 h Fig. 2b. The permutation test through 20 applications for this group and the other groups validated the PLS-DA model (Fig. 2c). Indeed, all Q2 values of permuted Y vectors were lower than the original ones and the regression of Q2 lines intersected the y-axis at below zero, indicating that the model was valid (Eriksson et al. 2001). A Mann–Whitney test was used to test the significance of the metabolites at the origin of the differential groupings. The corresponding discriminating metabolites were identified as lactate and glucose, and are visible in detail on the NMR spectra (Fig. 4a). The ratio of their corresponding areas showed an increase in lactate and a decrease in glucose when increasing the temperature and the pre-centrifugation delay. More specifically, the lactate content increased up to 2.67-fold and the glucose content decreased up to 1.67-fold between SER-SST-D-B-N-B-A and SER-SST-I-B-N-B-A (Table 3a).

Representative one dimensional NMR 1H spectrum obtained from a serum sample, with a CPMG presat

Score plot of principal component analysis, displaying data in the transformed coordinate space (a) and Partial Least Square analysis (b) based on the pre-centrifugation delay and temperature conditions. a (blue), 4 h RT; b (light blue), 4 h 4°C; c (red), 24 h RT; d (dark blue), 24 h 4°C. S1 to S7 represent samples from donors 1–7. Plots of permutation validation (c) of PLS-DA based on 1H-NMR spectra for the 24 h RT group (red) and the other groups (blue).The validation of the PLS-DA model by permutation tests through 20 applications resulted in a variance R2 of the model of 0.99 and a predictive ability Q2 of the model of 0.77
Samples from the same donor and of the same SPREC (SER-SST-C-B-N-B-A), having undergone 1, 5 or 10 freeze–thaw cycles, were then compared. We selected 3 PCs for the NMR spectra which explained 79.1% of the total variance but PCA analysis did not show any significant assemblage of samples (Fig. 3a). However, PLS-DA showed a significant grouping of samples which had undergone 5 or 10 freeze–thaw cycles in comparison with only one freeze–thaw cycle (Fig. 3b). A permutation test also validated this model (Fig. 3c). The quantitative analysis of the discriminating metabolites showed that decreases in choline, glycerol, methanol, ethanol, probably proline and in an unidentified peak at 1.91 ppm were at the origin of this differential grouping after 5 or 10 freeze–thaw cycles (Table 3b; Fig. 4b). As methanol and ethanol are volatile, multiple freeze–thaw cycles could explain their decrease through evaporation. Therefore, the common preanalytical variations linked to different pre-centrifugation conditions and/or the different numbers of freeze–thaw cycles had a visible impact on the metabolic profiles of serum samples. Targeted studies have shown no influence of such preanalytical variations on specific micromolecules such as cholesterol and micronutrients (Comstock et al. 2001). However, a global metabolomic approach revealed preanalytical profiles. Previous biospecimen research, examining the effect of the pre-centrifugation time and temperature and of freeze–thaw cycles on the metabolic profiles of serum, showed some slight alterations in the NMR profiles associated with longer pre-centrifugation delays. These alterations were mainly due to an increase in lactate. The pre-centrifugation delays studied by Teahan et al. (2006) were 0.5 versus 3 h. We studied longer delays, which are practiced more often in biobanks, and, especially in large scale epidemiological biobanks. The use of sodium fluoride or other glucose preservatives is not common practice in these biobanks. Therefore, our data confirm the increase in lactate, presumably due to continued anaerobic cell metabolism, and show that this increase continues at pre-centrifugation delays between 4 and 24 h. However, lactate does not increase if blood is stored at 4°C. This is compatible with the continued anaerobic metabolism hypothesis. Prolonged storage of blood at room temperature could also be seen by a decrease in the glucose zone. Lactate production and glucose consumption may be due to prolonged contact with erythrocytes which do not have mitochondria and can only produce energy by glycolytic fermentation of glucose to lactate. Alterations in low molecular weight metabolites were found to be associated with the repeated freeze–thaw of the serum. Some of the metabolites shown to decrease after repeated freeze–thaw, namely glycerol and choline, have been shown previously to be susceptible to post-centrifugation delays (Teahan et al. 2006). Metabolic profiles of human serum have been shown to be robust to post-centrifugation delays of up to 36 h (Barton et al. 2008). Metabolic profiles of rat plasma have also been shown to be robust to 9 month long storage at −80°C, although, increases in glycerol and choline were observed after storage of centrifuged plasma at room temperature or 4°C (Deprez et al. 2002). Finally, our results confirm results recently published by Bernini et al. (2011) showing glucose decrease and lactate increase with pre-centrifugation delays. Metabolites such as choline and proline which were shown to be sensitive to pre-centrifugation delays (Bernini et al. 2011), were also found to be sensitive to freeze–thaw cycles in the present study.

Score plot of principal component analysis, displaying data in the transformed coordinate space (a) and partial least square analysis (b) based on the number of freeze–thaw cycles. 1 day (blue), baseline 1 freeze–thaw cycle; 5 day (green), 5 freeze–thaw cycles; 10 day (dark green), 10 freeze–thaw cycles. S1 to S7 represent samples from donors 1–7. Plots of permutation validation (c) of PLS-DA based on 1H-NMR spectra for the 1 freeze–thaw cycle group (blue) and the other groups (green). The validation of the PLS-DA model by permutation tests through 20 applications resulted in a variance R2 of the model of 0.82 and a predictive ability Q2 of the model of 0.51

NMR spectra enlargements of the zones corresponding to the discriminating metabolites’ peaks for different pre-centrifugation delay and temperature conditions (a blue: 4 h RT; light blue: 4 h 4°C; red: 24 h RT; dark blue: 24 h 4°C) and different number of freeze–thaw cycles (b blue: 1 freeze–thaw cycle; green: 5 freeze–thaw cycles; dark green: 10 freeze–thaw cycles)
In conclusion, inter-individual variations play a major role in NMR spectroscopy profiles while specific preanalytical variations influence specific metabolites. Although more studies, including mass spectrometry, may be conducted to confirm these findings in biological fluids of other types and in solid tissues, these data suggest that serum is sensitive to certain preanalytical variations for proton NMR spectroscopic downstream applications. Standard Operating Procedures for preanalytical handling of blood for metabolomics studies have recently been proposed (Bernini et al. 2011). Systematic recording of the blood sample collection and preparation SOP details, according to a coding like SPREC (Betsou 2010) is recommended. This is in accordance with the previously published minimum requirements for designing and reporting metabolic studies (Lindon et al. 2005) in order to allow preanalytical factors to be included in subsequent multivariate analyses.
Abbreviations
- SST:
-
Serum separation tube
- SPREC:
-
Standard preanalytical code
- TSP:
-
3-(trimethylsilyl) 3,3,3,3-tetradeutero-propionic acid
- CPMG:
-
Carr-Purcell-Meiboom-Gill
- HSQC:
-
Heteronuclear single quantum coherence
References
Barton RH, Nicholson JK, Elliott P, Holmes E (2008) High-throughput 1H NMR-based metabolic analysis of human serum and urine for large-scale epidemiological studies/validation study. Int J Epidemiol 37 (Suppl 1):i31–i40
Bernini P, Bertini I, Luchinat C, Nincheri P, Staderini S, Turano P (2011) Standard operating procedures for pre-analytical handling of blood and urine for metabolomics studies and biobanks. J Biomol NMR 49:231–243
Betsou F, The ISBER Working Group on Biospecimen Science (2010) Standard preanalytical coding for biospecimens: defining the sample PREanalytical code (SPREC). Cancer Epidemiol Biomarkers Prevention 19:1004–1011
Comstock GW, Burke AE, Norkus EP, Gordon GB, Hoffman SC, Helzlsouer KJ (2001) Effects of repeated freeze-thaw cycles on concentrations of cholesterol, micronutrients, and hormones in human plasma and serum. Clin Chem 47:139–142
Deprez S, Sweatman BC, Connor SC, Haselden JN, Waterfield CJ (2002) Optimisation of collection, storage and preparation of rat plasma for 1H NMR spectroscopic analysis in toxicology studies to determine inherent variation in biochemical profiles. J Pharm Biomed Anal 30:1297–1310
Eriksson L, Johansson E, Kettaneh-Wold N, Wold S (2001) Multi- and megavariate data analysis: principles and applications. Umeå, Umetrics
Fujiwara M, Kobayashi T, Jomori T, Maruyama Y, Oka Y, Sekino H, Imai Y, Takeuchi K (2009) Pattern recognition analysis for 1H NMR spectra of plasma from hemodialysis patients. Anal Bioanal Chem 394:1655–1660
Gartland KP, Sanins SM, Nicholson JK, Sweatman BC, Beddell CR, Lindon JC (1990) Pattern recognition analysis of high resolution 1H NMR spectra of urine. A nonlinear mapping approach to the classification of toxicological data. NMR Biomed 3:166–172
Gavaghan CL, Holmes E, Lenz E, Wilson ID, Nicholson JK (2000) An NMR-based metabonomic approach to investigate the biochemical consequences of genetic strain differences: application to the C57BL10J and Alpk:ApfCD mouse. FEBS Lett 484:169–174
Goldsmith P, Raj Prasad K, Ahmad N, Fisher J (2009) 1H NMR spectroscopic study of blood serum for the assessment of liver function in liver transplant patients. J Gastrointestin Liver Dis 18:508–517
Griffin JL, Shockor JP (2004) Metabolic profiles of cancer cells. Nat Rev Cancer 4:551–564
He Q, Ren P, Kong X, Wu Y, Wu G, Li P, Hao F, Tang H, Blachier F, Yin Y (2011) Comparison of serum metabolite compositions between obese and lean growing pigs using an NMR-based metabonomic approach. J Nutrit Biochem (in press)
Holmes E, Nicholls AW, Lindon JC, Ramos S, Spraul M, Neidig P, Connor SC, Connelly J, Damment SJ, Haselden J, Nicholson JK (1998) Development of a model for classification of toxin–induced lesions using 1H NMR spectroscopy of urine combined with pattern recognition. NMR Biomed 11:235–244
Howe FA, Opstad KS (2003) 1H MR spectroscopy of brain tumours and masses. NMR Biomed 16:123–131
Kaartinen J, Hiltunen Y, Kovanen PT, Ala-Korpela M (1998) Application of self-organizing maps for the detection and classification of human blood plasma lipoprotein lipid profiles on the basis of 1H NMR spectroscopy data. NMR Biomed 11:168–176
Kriat M, Confort-Gouny S, Vion-Dury J, Sciaky M, Viout P, Cozzone PJ (1992) Quantitation of metabolites in human blood serum by proton magnetic resonance spectroscopy. A comparative study of the use of formate and tsP as concentration standards. NMR Biomed 5:179–184
Le Moyec L, Valensi P, Charniot JC, Hantz E, Albertini JP (2005) Serum 1H-nuclear magnetic spectroscopy followed by principal component analysis and hierarchical cluster analysis to demonstrate effects of statins on hyperlipidemic patients. NMR Biomed 18:421–429
Lindon JC, Holmes E, Nicholson JK (2001) Pattern recognition methods and applications in biomedical magnetic resonance. Prog Nucl Magn Reson Spectrosc 39:1–40
Lindon JC, Nicholson JK, Holmes E, Keun HC, Craig A, Pearce JTM, Bruce SJ, Hardy N, Sansone SA, Antti H, Jonsson P, Daykin C, Navarange M, Beger RD, Verheij ER, Amberg A, Baunsgaard D, Cantor GH, Lehman-McKeeman L, Earll M, Wold S, Johansson E, Haselden JN, Kramer K, Thomas C, Lindberg J, Schuppe-Koistinen I, Wilson ID, Reily Schaefer H, Spraul M (2005) Summary recommendations for standardization and reporting of metabolic analyses. Nat Biotechnol 23:833–838
Piotto M, Moussallieh FM, Dillmann B, Imperiale A, Neuville A, Brigand C, Bellocq JP, Elbayed K, Namer IJ (2008) Metabolic characterization of primary human colorectal cancers using high resolution magic angle spinning 1H magnetic resonance spectroscopy. Metabolomics 5:292–301
Psychogios N, Hau DD, Peng J, Guo AC, Mandal R, Bouatra S, Sinelnikov I, Krishnamurthy R, Eisner R, Gautam B, Young N, Xia J, Knox C, Dong E, Huang P, Hollander Z, Pedersen TL, Smith SR, Bamforth F, Greiner R, McManus B, Newman JW, Goodfriend T, Wishart DS (2011) The human serum metabolome. PLoS One 6(2):e16957
Sukumaran DK, Garcia E, Hua J, Tabaczynski W, Odunsi K, Andrews C, Szyperski T (2009) Standard operating procedure for metabonomic studies of blood serum and plasma samples using a 1H-NMR micro-flow probe. Magn Reson Chem 47:S81–S85
Teahan O, Gamble S, Holmes E, Waxman J, Nicholson JK, Bevan C, Keun HC (2006) Impact of analytical bias in metabonomic studies of human blood serum and plasma. Anal Chem 78:4307–4318
Tenenhaus M, Gauchi JP, Menardo C (1995) Regression PLS et applications. Revue Statistique Appliquée XLIII(1):7–63
Walsh MC, Brennan L, Malthouse JPG, Roche HM, Gibney MJ (2006) Effect of acute dietary standardization on the urinary, plasma, and salivary metabolomic profiles of healthy humans. Am J Clin Nutr 84:531–539
Wishart DS, Knox C, Guo AC et al. (2009) HMDB: a knowledgebase for the human metabolome. Nucleic Acids Res 37(Database issue):D603–D610
Acknowledgments
This work was supported by the Conseil Régional de Picardie. We are grateful to Brian de Witt and Karsten Hiller for critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Fliniaux, O., Gaillard, G., Lion, A. et al. Influence of common preanalytical variations on the metabolic profile of serum samples in biobanks. J Biomol NMR 51, 457–465 (2011). https://doi.org/10.1007/s10858-011-9574-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-011-9574-5