
Abstract
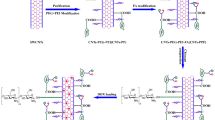
A novel dual-drug delivery system (DDDS) for cancer chemotherapy has been established by employing highly purified and mildly oxidized large-inner-diameter multi-walled carbon nanotubes (LID-MWCNTs) as the vector. The LID-MWCNTs were modified with the antitumor drugs, cisplatin (CDDP) and doxorubicin (DOX). CDDP was encapsulated inside the nanotube vectors by a wet-chemical approach while DOX was attached to the external surfaces through non-covalently interaction. The loading efficiencies of CDDP and DOX were as high as 84.56 and 192.67%, respectively. Notably, after CDDP was encapsulated inside the nanotubes, a three-level blocking strategy, which included polyethylene glycol, folic acid and DOX, was employed to block the CDDP exits at different levels. The pH-sensitive release profile of CDDP was demonstrated using a modified characterization method, as well as that of DOX. Finally, the anticancer activity of the DDDS on MCF-7 cells was tested and a synergistic effect was recorded. This work is part of our LID-MWCNTs based drug delivery system studies, and provides a basis for developing a novel comprehensive antitumor treatment that combines chemotherapy and photothermal therapy.
Graphical Abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Cancer is one of the most dreadful and devastating diseases. So far, many anticancer agents have been developed. However, repeated administration of a single kind of drug is often accompanied with detrimental side effects, including intolerable cytotoxicity, non-targeting, and resistance to chemotherapies, among others [1–3]. To address the hurdles associated with conventional single drug treatment, the concept of combination drug therapy has been employed [4, 5]. Not only does combination drug therapy exert a synergistic effect to enhance the pharmacological action, but also reduces individual drug-related toxicity [1, 6, 7]. However, current combination drug therapy is far from perfect. Poor solubility of anticancer agents and their low ability to be transported across physiological barriers often lead to undesirable biodistribution, which seriously compromises its potential advantages in clinical applications. Additionally, employing two separate drug delivery systems increases the difficulty in clinical operation and the high dose of the vector might exhibit a considerable adverse effect on normal tissue. Therefore, the need for developing a dual-drug delivery system (DDDS) that can simultaneously transport both drugs to the tumor sites, as well as to provide prolonged release kinetics is urgently needed to secure the therapeutic benefits of co-delivery of drugs.
Among various vectors in drug delivery systems, carbon nanotubes (CNTs) have attracted attention for their nanoneedle shape, hollow structure, and excellent chemical properties [8]. Anticancer agents can be loaded either outside or inside CNTs. The prerequisite for improving drug loading efficiency of CNT based drug delivery systems is that CNT carriers possess a huge inner space and a large surface area. Currently, a novel CNT family member, the thin wall large-inner diameter (20~50 nm) multi-walled carbon nanotubes (LID-MWCNTs) which have already attracted interest in other fields [9], might be considered as promising vectors to improve drug loading efficiency. Hence, LID-MWCNTs were employed to establish DDDS in the current study.
Loading a single kind of anticancer agent (either outside or inside) onto CNTs has already been investigated [10–12]. However, more than one drug to CNTs at the same time is a frontier of research and has rarely been reported. Theoretically, attaching different drugs to the external surface of nanotubes is feasible. However, it has been predicted that the binding procedure may reduce the individual drug doses, particularly when different loading mechanisms involve a common reaction or binding site. Meanwhile, given that only a few kinds of drugs can be encapsulated inside the nanotubes, incorporating two drugs with different antitumor mechanisms has also been difficult. To address the deficiencies mentioned above, we designed a LID-MWCNTs based DDDS in which doxorubicin (DOX) was attached to the external surfaces while cisplatin (CDDP) was encapsulated inside the inner cavities of the nanotubes. Additionally, with the cargo drugs separated by the sidewall of the nanotubes, the space of the CNTs could be fully occupied, which was more conducive to further improvements to the loading efficiency of both drugs.
When designing drug delivery systems, the release profile of the cargo drug is a key issue to be considered. Facing with the fact that the pH of tumor extracellular matrix (pH 6.5–7.0) is lower than that of normal tissue (pH 7.4) [13, 14], Establishing the pH-sensitive drug delivery system is considered to be a valuable strategy with practical implications [15, 16]. The pH-sensitive DDDS could diminish the early release of cargo drugs during circulation in blood, and consequently could ensure that more drug would be delivered to the tumor tissue. It has been fully demonstrated that external-surface-loaded non-covalently-bonded DOX undergoes pH-sensitive release [17]. Therefore, the core issue in building the novel DDDSs was how to achieve the pH-sensitive release profile of encapsulated CDDP. The technological difficulty mainly existed in finding the suitable pH-detachable blocker that can alter the size of the CDDP exit in different pH environments, thereby controlling the CDDP release. Based on the principle of simplifying the construction process and endowing the DDDS with potential multifunctional properties, we applied a three-level blocking strategy to tentatively block the CDDP exit at different levels by using three commonly-used bioactive molecules—polyethylene glycol (PEG), folic acid (FA) and DOX.
The present research describes our attempt to develop a novel LID-MWCNT based pH-sensitive DDDS. Prior to loading drugs, the highly purified and low oxidized LID-MWCNTs (O-LID-MWCNTs) were prepared. Then, the CDDP encapsulation and PEG grafting (i.e. primary blocking procedure) were carried out in sequence. The resulting product (CDDP@O-LID-MWCNTs-PEG) was subsequently subjected to FA bonding (i.e. secondary blocking procedure) and DOX attachment (i.e. tertiary blocking procedure), followed by the characterization of loading efficiency and release profile of this novel DDDS. Additionally, the cytotoxicities toward mouse fibroblast cells and breast cancer cells were investigated in vitro. To the best of our knowledge, this is the first time that CNTs have been employed as vectors to construct DDDS. More importantly, besides significantly improving the loading efficiency of both drugs, we successfully proposed and validated the three-level blocking hypothesis that DOX can trigger the pH-sensitive release of CDDP by simply blocking the exit of CDDP with PEG, FA, and DOX. And the pH-sensitive release pattern of both drugs was finally obtained.
2 Materials and methods
2.1 Synthesis of FA bonded CDDP@O-LID-MWCNTs-PEG
CDDP@O-LID-MWCNTs-PEG was synthesized according to the previous method [18] with modification (the detail method was shown in online resource: Materials and methods). To introduce the FA as the secondary blocker, 15 ml of dimethyl formamide (DMF) (Sigma-Aldrich, St. Louis, MO, USA) solution of CDDP@O-LID-MWCNTs-PEG was prepared before adding 10 mg FA (Sigma-Aldrich, St. Louis, MO, USA), 5 mg N,N-diisopropyl-ethylamine, 4-(methylamino) pyridine (DMAP) (Sigma-Aldrich, St. Louis, MO, USA) and 5 mg N-(3-dimethylaminopropyl)-N-ethylcarbodiimideHCl (EDC.HCl) (Sigma-Aldrich, St. Louis, MO, USA). After stirring at room temperature in darkness for 12 h, the solution was microfiltered through a 0.1 μm pore size polytetrafluoroethylene (PTFE) membrane (Millipore, Bedford, MA, USA) with repeated washing. The product (CDDP@O-LID-MWCNTs-PEG-FA) was measured by UV–VIS spectroscopy (UV-3600, SHIMADZU, Kyoto, Japan).
2.2 Loading DOX onto CDDP@O-LID-MWCNTs-PEG-FA
Loading DOX onto CDDP@O-LID-MWCNTs-PEG-FA was performed according to a modification of the previously described method [12]. Briefly, 3 mg DOX hydrochloride (Sigma-Aldrich, St. Louis, MO, USA) and 3 mg CDDP were dissolved in 15 ml of phosphate buffer solution (PBS) (pH 7.4) by sonication in darkness. Then, the CDDP@O-LID-MWCNTs-PEG-FA containing 1 mg O-LID-MWCNTs was added into the solution and stirred for 6 h at room temperature in darkness. After this procedure, DOX was loaded not only to the external surface of the nanotube vectors, but also to the secondary blocker molecules (i.e. FA). Thus, the three-level blocking was constructed and the entrapped CDDP was almost completely sealed inside nanotube vectors. The resulting DDDS, denoted as CDDP@O-LID-MWCNTs-PEG-FA-DOX, were collected by repeated ultrafiltration (MWCO = 10 kDa) and were thoroughly washed with PBS until the supernatant became colorless and then they were freeze-dried. The unbounded DOX was collected and determined by measuring the UV absorbance at 488 nm (the characteristic absorbance of DOX) relative to a calibration curve recorded under identical conditions, allowing the DOX loading efficiency to be estimated using following Eq:
The microstructures of CDDP@O-LID-MWCNTs-PEG-FA-DOX were observed by scanning electron microscopy (SEM) (Inspect F50, FEI, Hillsboro, OR, USA) and high-resolution transmission electron microscopy (HR-TEM) (FEI, Hillsboro, OR, USA). The anchoring of DOX along the external surface of O-LID-MWCNTs was confirmed by UV–VIS spectroscopy. Additionally, the CDDP loading efficiency of DDDS was re-measured by the previous method (the measurement method of CDDP loading efficiency was shown in online resource: Materials and methods).
Meanwhile, FA non-bonded DDDS (CDDP@O-LID-MWCNTs-PEG-DOX) was also prepared using CDDP@O-LID-MWCNTs-PEG by the same method. Its microstructure was investigated by SEM and HR-TEM and the DOX loading efficiency was measured.
2.3 In vitro drug release study
DOX release profiles were investigated according to the dialysis method [12]. FA bonded DDDS was suspended in 5.0 ml of PBS buffer (pH 7.4 and 6.5) and sealed in a dialysis bag (MWCO = 1 kDa). The bag was immersed in 45 ml of PBS (pH 7.4 and 6.5) in darkness. At each time interval, 10 ml of the dialysate was removed to determine the concentration of the released DOX by UV–VIS spectroscopy, and then was added back to the original PBS. The experiments were performed in triplicates.
Since organic compounds like DOX may affect the accuracy of profile spec inductively coupled plasma optical emission spectrometer (ICP-OES) results, the release profiles of CDDP were determined by a different method. Briefly, 12 parallel samples of CDDP@O-LID-MWCNTs-PEG-FA-DOX were prepared. Then, each sample was dispersed in 5.0 ml of PBS buffer (pH 7.4 and 6.5) and sealed in a dialysis bag (MWCO = 1 kDa). Each bag was submerged into 45 ml of one of the corresponding PBS solutions at 37 °C in darkness. At each time intervals, one of the parallel samples was subjected to the following analysis. The suspension in the dialysis bag was taken out and microfiltered through a 0.1 mm PTFE membrane. Then it was dried in an oven at 100 °C. The resulting powders were heated at the rate of 30 °C min−1 to a final temperature of 1000 °C and kept at that temperature for 1 h in an air atmosphere. The residue was dissolved in diluted aqua regia and analyzed by ICP-OES (Teledyne Leeman Labs, Hudson, NH, USA) to determine the amount of remaining CDDP, which represented the dose of unreleased CDDP. Given the known initial dose of encapsulated CDDP, we could calculate the dose of released CDDP at each time point. In this way, the CDDP release profiles were obtained.
To verify the effect of secondary and tertiary blockers on the drug release property, the CDDP release profiles of CDDP@O-LID-MWCNTs-PEG-DOX were also determined using the same method.
2.4 In vitro cytotoxicity assay
The cytotoxic activity of empty vector was evaluated against the mouse fibroblast L929 cells and in the human breast cancer cell line MCF-7 supplied by State Key Laboratory of Oral Diseases (Chengdu, China). The antitumor efficacy of DDDS was tested using MCF-7. The L929 and MCF-7 cells were cultured and grown in Dulbecco’s modified Eagle medium (DMEM) and 1640 Medium (Gibco, Life Technology, Grand Island, NY, USA), respectively, supplemented with 10% fetal bovine serum (FBS) (Atlanta Biologicals, Lawrenceville, GA) and 1% penicillin–streptomycin (P/S) (Gibco, Life Technology, Grand Island, NY, USA) at 37 °C under a humidifying atmosphere containing 5% CO2 and a 21% O2. All tests were performed on cells in the logarithmic phase of growth.
To evaluate the cytotoxic activity of empty vectors, the cytotoxicity assays of O-LID-MWCNTs, O-LID-MWCNTs-PEG, and O-LID-MWCNTs-PEG-FA were conducted on L929 and MCF-7. Additionally, to verify the antitumor efficacy of DDDS on MCF-7, the free DOX solutions were diluted with culture medium to obtain five different concentrations (1.25–20 μg/ml). Then, equivalent DOX concentrations of CDDP@O-LID-MWCNTs-PEG-FA-DOX and O-LID-MWCNTs-PEG-FA-DOX solutions were prepared based on the respective drug loading efficiencies. According to the amount of CDDP in CDDP@O-LID-MWCNTs-PEG-FA-DOX, the equivalent concentrations of free CDDP and CDDP@O-LID-MWCNTs-PEG-FA were also prepared. The free CDDP was mixed with free DOX solution. The combination of free drugs (CDDP + DOX), the nanotube based single drug delivery systems (O-LID-MWCNTs-PEG-FA-DOX and CDDP@O-LID-MWCNTs-PEG-FA), and the FA-bonded DDDS were tested. Cytotoxicity assays were conducted by the Cell Counting Kit-8 (CCK-8) assay [19, 20]. Briefly, L929 and MCF-7 cells were seeded onto 96-well plates at a density of 104 cells well−1. After cell adhesion, the medium was replaced by 150 μl of fresh medium which contained empty vectors, free drugs, cargo drugs, or DDDS (pH 7.4 and 6.5). Then, the cells were incubated at 37 °C in 5% CO2 and were measured at various durations, including 24, 48, and 72 h. At each time interval, the medium was removed and the wells were washed three times with PBS before 10 μl of CCK-8 solution was added and incubated with cells for 3 h. The optical density (OD) at 450 nm was measured by the PowerWave™ 340 microplate spectrophotometer (BioTek, Winooski, VT, USA). Cells without any treatment were considered as negative controls. Triplicate samples from each group were used to assess the cell viability.
2.5 Statistical analysis
Statistical analyses were performed with SPSS-PC17.0 Software (SPSS Inc., Chicago, Illinois, USA). In vitro cell viability values (OD) were statistically compared using a one-way ANOVA and LSD tests. The level of significance for all statistical analyses was defined as p < .05.
3 Results
3.1 Synthesis of CDDP@O-LID-MWCNTs-PEG
Contrary to the micromorphologies of pristine LID-MWCNTs, almost all the impurities were removed and only a few defects were observed along the external surface of O-LID-MWCNTs (Fig. S1). The appearance of C=O peak (around 1723 cm−1) in FTIR (Fig. S2a) demonstrated the effective oxidation of O-LID-MWCNTs and the residual metal catalyst mass plunged to 2.94 wt% (Fig. S2b). Additionally, no Pt element was detected in the O-LID-MWCNTs.
Investigated by HR-TEM and EDX (Fig. S3), numerous CDDP particles were entrapped inside the O-LID-MWCNTs and tended to agglomerate. By grafting to the open-ends and sidewall defects of nanotubes, the semilunar-shaped PEG could serve as the primary blocker to control the release of entrapped CDDP. The successful grafting of PEG to O-LID-MWCNTs was also confirm by the FTIR spectrum (Fig. S4a). Moreover, free CDDP had been completely removed from the external surfaces of CDDP@O-LID-MWCNTs-PEG due to no characteristic peak of CDDP was detected in the RAMAN spectrum of CDDP@O-LID-MWCNTs-PEG (Fig. S4b).
3.2 Characterization of FA bonded and FA non-bonded DDDS
Compared with SEM results of the vectors (O-LID-MWCNTs) in Fig. S1b, the diameter of DDDS was significantly increased and its external surface appeared to be rather ‘rough’ (Fig. 1a and c). Additionally, although the DDDS was dramatically thickened and appeared to be less well defined in contrast with the HR-TEM result for CDDP@O-LID-MWCNTs-PEG, many CDDP particles still remained inside the nanotubes (Fig. 1b and d). Interestingly, the microstructures of FA bonded DDDS and its counterpart were entirely different. In the FA non-bonded DDDS (Fig. 1a), the attached substances were irregularly distributed along the vectors, leaving many sites not fully covered. This phenomenon was particularly clear in HR-TEM (Fig. 1b) which showed the uncovered sites to be scattered along the axis of the FA non-bonded DDDS. Meanwhile, within the corresponding side of nanotube, we observed that the encapsulated CDDP was centrally distributed. Noticeably, some CDDP particles were just released from the nanotube and passed through the uncovered sites, which indicated these sites may be the exit site for encapsulated CDDP. However, the sludge-like attached substances were evenly distributed along the external surface of vector both in the SEM and HR-TEM images in FA-bonded DDDS. Similarly, the trend of the centralized distribution of CDDP was also found.

The microstructure of FA non-bonded and FA bonded DDDS. a SEM and b HR-TEM images of CDDP@O-LID-MWCNTs-PEG-DOX. c SEM and d HR-TEM images of CDDP@O-LID-MWCNTs-PEG-FA-DOX. The white arrows in Fig. 5b mark the sidewall defects along the external surface of O-LID-MWCNTs not fully covered by DOX
We observed the characteristic peak of FA (282 nm) in the UV–Vis spectra of FA-bonded DDDS and CDDP@O-LID-MWCNTs-PEG-FA (Fig. 2). However, this peak was slightly blue-shifted, indicating chemical interactions had occurred between PEG and FA [21]. The peak of DOX appears at 490 nm. When DOX interacted with CDDP@O-LID-MWCNTs-PEG-FA and CDDP@O-LID-MWCNTs-PEG, the DOX peak showed a red-shift (499 nm). The simultaneous emergence of the characteristic peaks of FA and DOX in FA-bonded DDDS indicated the successful grafting of FA and DOX to nanotubes.

The UV–vis absorption spectra of FA, DOX, CDDP@O-LID-MWCNTs-PEG, CDDP@O-LID-MWCNTs-PEG-FA, CDDP@O-LID-MWCNTs-PEG-DOX and CDDP@O-LID-MWCNTs-PEG-FA-DOX
3.3 The drug loading efficiency and drug release profile
The loading efficiencies of the drugs are shown in Table 1. A high CDDP loading efficiency of 92.80% for CDDP@O-LID-MWCNTs-PEG was achieved. Although the encapsulated CDDP amount decreased after the FA bonding and DOX loading procedures, the loading efficiency of 84.56% could still be obtained for FA-bonded DDDS. As for DOX, the loading efficiency of FA-bonded DDDS (192.67%) was increased by 18.60% compared with that of the FA non-bonded DDDS (174.07%).
The CDDP release profiles of CDDP@O-LID-MWCNTs-PEG, FA non-bonded DDDS, and FA-bonded DDDS are presented in Fig. 3. Except for FA-bonded DDDS at pH 7.4, which exhibited a shorter burst release and a relatively short sustained release, all other five release profiles were characterized by a shorter burst release phase and a longer sustained release phase. Notably, the release profiles at different pH values in FA-bonded DDDS were entirely different from those in CDDP@O-LID-MWCNTs-PEG and FA non-bonded DDDS. The CDDP release amount of FA-bonded DDDS was distinguish from that of CDDP@O-LID-MWCNTs-PEG and FA non-bonded DDDS. For example, T10% (time needed to release 10% of CDDP) is around 0.5 h for CDDP@O-LID-MWCNTs-PEG and FA non-bonded DDDS under both pH condition, whereas T10% increased to 4.7 and 6.5 h for FA-bonded DDDS at pH 6.5 and 7.4, respectively. Additionally, in the release profile of CDDP@O-LID-MWCNTs-PEG and FA non-bonded DDDS, the pH-sensitive release pattern was barely detected. In FA-bonded DDDS, however, the released amount of CDDP at pH 6.5 was significantly increased compared with that at pH 7.4.

The release profiles of CDDP from CDDP@O-LID-MWCNTs-PEG, CDDP@O-LID-MWCNTs-PEG-DOX, and CDDP@O-LID-MWCNTs-PEG-FA-DOX at different pH values
Figure 4 exhibits the DOX and CDDP release profile of FA-bonded DDDS. Although the burst release phase and cumulative release amount was relatively low in pH 7.4 compared with those of in pH 6.5, the biphasic release profile of cargo drugs was observed in FA-bonded DDDS under different pH conditions, and as predicted, the pH-sensitive release pattern of DOX was observed. Similarly, a strong pH-sensitive release profile for CDDP was also demonstrated in FA-bonded DDDS. The CDDP cumulative release in 72 h amounted to 13 and 26% at pH 7.4 and 6.5, respectively. Interestingly, the release profile of CDDP was comparable with that of DOX under the same pH conditions. For instance, the release of CDDP quickly reached equilibration at pH 7.4 after the burst release, and almost no sustained release phase was detected. In contrast, a sustained release of CDDP continued over an extended period and did not reach equilibration in 72 h at pH 6.5. In addition, the similarity factor, f2, was also used to compare the similarity of DOX and CDDP release profile under the same pH condition. F2 for DOX and CDDP release profile was 68.8 and 71.2 at pH 7.4 and 6.5, respectively. The value of similarity factor (f2 > 50) confirmed that the release profile of CDDP was similar to that of DOX under the same pH conditions. To further exploit the relationship between the release profiles of the two drugs, the release patterns of DOX and CDDP at the early stages (1–12 h) was determined, as indicated in Fig. 4c and d, respectively. For the DOX (Fig. 4c), an initial burst of drug release gradually gave way to a slower rate of drug release at both pH values. For CDDP (Fig. 4d), the release rate was relatively low within the first 2 h before a surge of drug release occurred. Then, the drug release behaved with the same release profile as DOX displayed under the same pH environments.

The release profiles of a DOX and b CDDP from CDDP@O-LID-MWCNTs-PEG-FA-DOX at different pH values. The release profiles in the early stages for DOX and CDDP are shown in c and d, respectively
3.4 Cytotoxicity
We investigated the cytotoxicity of the empty vectors (O-LID-MWCNTs, O-LID-MWCNTs-PEG, and O-LID-MWCNTs-PEG-FA) in L929 and MCF-7 cells (Fig. S5). In the present study, the loading efficiency of each drug delivery system was entirely different and thus, we had to prepare and measure many concentrations of empty vectors. To simplify the test procedure, we selected a broad range of concentrations of empty vectors, which covered all the doses that needed to be tested. As seen in Fig. S5, all the empty vectors exhibited a low toxicity and a remarkable biocompatibility in tumor and non-tumor cells (cells viabilities >95%) at various concentration.
The antitumor efficacy of FA-bonded DDDS under pH 7.4 was evaluated against MCF-7 (Fig. 5a–d), whose proliferation was sensitive to DOX treatment, at 24, 48, and 72 h, and its efficacy was compared with that of O-LID-MWCNTs-PEG-FA-DOX, CDDP@O-LID-MWCNTs-PEG-FA, and the free DOX and CDDP groups. The tumor cells displayed a concentration-dependent decrease in viability when exposed to the four group treatments. In the O-LID-MWCNTs-PEG-FA-DOX (Fig. 5a) group, MCF-7 exhibited a strong time-dependent effect, which may result from the sustained release of loaded DOX. On the other hand, since almost all the encapsulated CDDP were released during the first 24 h in the CDDP@O-LID-MWCNTs-PEG-FA group (Fig. 5b), MCF-7 did not significantly decrease in the following 48 and 72 h. This phenomenon was particularly clear when the concentration was lower than 5 μg/ml. Due to an absence of sustained release, the proliferation of tumor cells was severely decreased in the first 24 h in the free DOX and CDDP group (Fig. 5c), and the cells did not show a time-dependent decrease in cell viability in the subsequent incubation. For the FA-bonded DDDS group (Fig. 5d), a remarkable synergistic effect could be found at all concentration and time points comparing the antitumor efficacy with that of the O-LID-MWCNTs-PEG-FA-DOX group (Fig. 5a) and CDDP@O-LID-MWCNTs-PEG-FA group (Fig. 5b). However, the antitumor efficacy was significantly lower than that in the free DOX and CDDP group (Fig. 5c) at each concentration in the first 24 h. Then, owing to the sustained release of both drugs, the DDDS started to exhibit an exciting antitumor efficacy during the following incubation. At the time point of 72 h, the DDDS group showed more cytotoxicity than that in the free DOX and CDDP group, which was particularly obvious at the high concentration (≥5 μg/ml). Moreover, the cytotoxicity of DDDS on MCF-7 cells under pH 6.5 was tested to mimicking the weak acidic microenvironment of tumor (Fig. 5e). The antitumor efficacy of DDDS under pH 6.5 was higher than that under pH 7.4, indicating that the drug release was enhanced in acidic environment. The antitumor efficacy of DDDS on MCF-7 cells was also investigated by phase-contrast microscopy and the images were shown in Fig. S6. With the incubation time increasing, the DDDS induced cell growth inhibition gradually gave way to cell death, which may be related with the synergistic antitumor effects of CDDP and DOX. Relatively, a more pronounced antitumor effect was observed in the pH 6.5 condition than that in pH 7.4 condition in day 3.

The viability studies of MCF-7 cells after incubation with a O-LID-MWCNTs-PEG-FA-DOX, b CDDP@O-LID-MWCNTs-PEG-FA, c free DOX and CDDP, d CDDP@O-LID-MWCNTs-PEG-FA-DOX at pH 7.4 and e CDDP@O-LID-MWCNTs-PEG-FA-DOX at pH 6.5. The cellular viability was calculated as a percentage of the viability of the untreated cells
4 Discussion
Co-delivery of different drugs for the treatment of cancer has attracted increasing attention because of the synergistic antitumor effects and inhibition of multidrug resistance. So far, a series of vectors including liposome [22], dendrimer [23] and polymeric micelle [24] based DDDSs have been explored. However, as far as our information goes, CNT based DDDS were seldom studied and rarely reported. The reason may be concerned with the technological difficulties with the following factors: the accurate control of the complicated physical-chemical properties of the vector (carbon nanotubes), the design of a suitable loading sequence for different drugs, the avoidance of undesired interference between the drugs and other bioactive molecules, solving the problem of low loading efficiency of both drugs, obtaining a sustained and pH-sensitive release profile for both drugs and the precise characterization of the CNT based DDDS. To mitigate these technological difficulties, we employed the mildly oxidized LID-MWCNTs as the vector to construct a novel DDDS characterized by high loading efficiency of both drugs and making the maximum utilization of the space of nanotubes, where CDDP was entrapped inside the cavity of the nanotubes and DOX was attached to the external surface. More importantly, since the release path of encapsulated CDDP was somewhat blocked by the attached DOX, the variations in the release pattern of DOX under different pH condition could correspondingly alter the release pattern of CDDP. Consequently, both CDDP and DOX exhibited a pH-sensitive release pattern.
As confirmed by TGA, SEM, and FTIR (Figs. S1, 2), after the purification and mild oxidation procedure, the overwhelming majority of impurities in pristine LID-MWCNTs (carbonaceous impurities, particles, and metal catalysts) were removed, and the carboxyl groups were introduced to these highly purified O-LID-MWCNTs, which led to a better dispersibility and provided the foundation for further PEG grafting.
According to the published literature [25, 26], the open ends and the sidewall defects along the oxidized nanotubes can act as the paths for CDDP to enter and then be released. Since a single nanotube vector only has two open ends at most, the number of sidewall defects contributes significantly to the quantity of paths of CDDP. Theoretically, a large number of sidewall defects could facilitate entry of CDDP and improve CDDP loading efficiency within a short encapsulation time. However, an increase in the number of sidewall defects could undoubtedly increase the difficulty in regulating the release pattern of CDDP. Considering the fact that the large inner space of LID-MWCNTs could originally enhance the loading efficiency of CDDP and summarizing the balance between harm and benefit brought about by decreasing the number of sidewall defects, we selected a mild oxidation method by decreasing the oxidation reaction intensity (lowering the temperature of reflux and rate of agitation) during the H2SO4/HNO3 reflux. As demonstrated by the results, only a few sidewall defects were observed along the O-LID-MWCNTs, an outcome that was entirely different from the previously reported oxidized nanotubes [18, 27]. Moreover, to guarantee a certain amount of DOX loading sites (through π-π stacking) along the external surface and to ensure the large inner volume of the single nanotube, we selected the nanotubes that were longer than 450 nm by microfiltration. Meanwhile, the resulting O-LID-MWCNTs were subjected to repeated washing with the goal of eliminating the ultra-short nanotubes [28] that might attach to the surface of O-LID-MWCNTs and further affect the subsequent blocking procedure.
Designing a suitable loading sequence of drugs is an essential aspect for building DDDSs. In this study, non-covalently bonded DOX was attached via the external-surface-loading approach, whereas CDDP was entrapped via the inner-cavity-loading approach where capillarity is the driving force. Theoretically, if the external-surface-loading procedure is implemented before the inner-cavity-loading procedure, the attached DOX will increase the length of entry path for CDDP as well as narrow its width, which will make CDDP encapsulation more difficult. Hence, the inner-cavity-loading procedure was conducted prior to external-surface-loading procedure in our methodology.
The release profile is also a fundamental parameter for drug delivery systems. However, prior to analyzing the release pattern of drugs, the measurement method is a critical issue. In the previous investigations where CDDP is the only drug in a CNTs-based drug delivery system [29], the common method to obtain the release profile is to simply employ a dialysis method in combination with ICP-OES measurement. Typically, an aliquot of dialysate containing the released CDDP is periodically collected during the dialysis procedure and is directly measured by ICP-OES. After calculating the proportion of released dose and accounting for the total encapsulated dose, the release profile of CDDP may be obtained. However, both attached DOX and encapsulated CDDP are released at the same time in our multicomponent DDDS, and the DOX-containing dialysate might affect the accuracy of ICP-OES results. In order to eliminate the adverse effect caused by the presence of organics, we modified the measurement method (described in Section 2.5). Briefly, the DDDS that contained unreleased CDDP were taken out instead of the dialysate. After microfiltration, drying, and calcination procedures, all the organic components in DDDS were removed and the residue containing unreleased CDDP was measured by ICP-OES. Subsequently, the amount of released CDDP was calculated based on the initial dose of encapsulated CDDP and the release profile was obtained. We compared the CDDP release profile of CDDP@O-LID-MWCNTs-PEG (Fig. 3) with that measured by the common method in our previous study [18], and noted that the two results were comparable to each other, which demonstrates the reliability of this modified measurement method. Moreover, since the release dose of CDDP is usually extremely low at the very early stage, even below the detection range of ICP-OES, we assumed that the modified measurement method, which measures the high-dosed unreleased CDDP, was more advantageous.
As is known to all, the extracellular matrix of tumor is weak acidic with pH around 6.5–7.0, which is lower than that of normal tissue (pH 7.4) [14]. Hence, the pH difference between normal tissue and tumor tissue could be employed as a triggering mechanism of specificity for DDDS. Correspondingly, obtaining the pH-sensitive release and sustained pattern was the key issue. In our study, DOX was designed to attach to the external surface of the vector through non-covalent bonding forces that could become weakened under acidic conditions, and as confirmed by our results, both pH-sensitive and a sustained release profile were obtained for the attached DOX. As for CDDP, a similar strategy of weakening the interaction between the drug and the nanotubes in low pH seemed impossible, because it was demonstrated that there is no interaction between the encapsulated CDDP and the internal surface of the nanotubes [25]. The literature, including our own study, revealed that CDDP encapsulated in unblocked nanotube vectors would exhibit a rapid release. Furthermore, it was suggested that the only way to control the release pattern of entrapped CDDP was to regulate the size of its exits (open ends and sidewall defects in nanotubes) [18, 25, 28]. Considering the size of the exits in O-LID-MWCNTs, high molecular weight PEG was grafted onto the O-LID-MWCNTs to initially block the exits. With this primary blocking, massive drug loss would be avoided during the initial burst release phase and a sustained release profile could be achieved. Nevertheless, the release of CDDP was barely affected by the pH after primary blocking. Therefore, we need to further block the exits for CDDP to achieve pH-sensitive release profiles.
When selecting the secondary blocker, there are several requirements to consider. First, the blocker should be attached to the surface of already grafted PEG (primary blocker). Second, the blocker should not generate undesired side effects in either normal tissue or on the nanotubes. Third, the blocker should not occupy the DOX binding sites and subsequently decrease the loading efficiency of DOX. Last but not least, the secondary blocker should detach from the primary blockers under acidic pH conditions in order to achieve a pH-sensitive release of incorporated CDDP. Interestingly, as aforementioned, non-covalent bonded DOX presents an acidic pH-sensitive release property. Hypothetically, if DOX was involved as a part of the blockers, all the requirements mentioned above could be fulfilled. Unfortunately, as demonstrated by the previous studies [30] and our preliminary experiments, DOX could not directly attach to the surface of PEG, and consequently could not serve as the secondary blocker.
Notably, Niu et al. reported an increased DOX loading efficiency of 13% for FA-conjugated PEGylated CNTs compared to its counterpart without FA conjugation, while the DOX release profile remained the same. The authors attributed the increased DOX loading to the electrostatic interaction between FA and DOX [31]. In a similar study [32], Huang et al. confirmed that an additional hydrogen bond existed between FA and DOX in a CNT based drug delivery system. Those studies suggested the potential of FA serving as the ‘bridge region’ between PEG and DOX. And the biocompatibility of FA has been fully demonstrated by numerous reports [33]. Therefore, we employed FA as the secondary blocker in this study. On one hand, FA was covalently bonded to PEG primary blocker. On the other hand, it was non-covalently combined with DOX which served as the tertiary blocker and controlled the release of CDDP in a pH-sensitive way.
Apart from their respective roles in controlling the release of CDDP, each component in the three-level blocking strategy may have its own additional function. The primary blocker, PEG, not only endows the nanotubes with better dispensability and excellent biocompatibility, but also extends the circulation period of the nanotube vectors by protecting them from being eliminated by the reticulo-endothelial system [34, 35]. The secondary blocker, FA, theoretically might play a targeting role in facilitate the DDDS entering the cells via endocytosis [17, 32, 36, 37], although this function was currently affected by DOX (acting as tertiary blocker) as well as the esterification reaction with PEG in this study. As for the tertiary blocker, there is no difference in antitumor effect between the blocker DOX and the external-surface-loaded DOX. Both species could generate a synergistic effect in combination with CDDP.
After the CDDP exit was blocked by PEG, FA and DOX, the CDDP was almost completely sealed inside nanotube. Since the release of non-covalently-bonded DOX was rather weak under neutral condition, the CDDP exit remained highly blocked and the overwhelming majority of CDDP cannot be release. However, when exposed to the acidic condition, numerous tertiary blocker of DDDS would detach from the FA and its blocking effect on exit was impaired. With sustained releasing of DOX, the blocked CDDP exit continues to be enlarged and encapsulated CDDP correspondingly released. Thereby, CDDP could exhibit the pH-sensitive release profile.
With the three-level blocking strategy, the incorporated CDDP presents a pH-sensitive release profile. Its 72 h cumulative release at pH 5.5 was 26% while that at pH 7.4 was only 13%. Similarly, the 72 h cumulative release of DOX at pH 5.5 and pH 7.4 were 22 and 8%, respectively. The pH-sensitive release profiles indicated that the DDDS would release only a few drugs during transportation in body, while an accelerated release could be triggered when it reached the acidic tumor tissues. Notably, the burst release of CDDP started 2 h later than that of DOX. It is probably because the released amount of DOX blockers within the first 2 h was limited, and the sizes of the CDDP exits were too small for the majority of encapsulated CDDP to be released. With continuing release of DOX, the size of CDDP exits became enlarged, which correspondingly promoted the release of CDDP. After that, the comparable release trend of the two drugs would exert a synergistically antitumor effect.
Besides the release profile, the drug loading efficiency is another key issue for DDDSs. High loading efficiency signified that we can obtain clinical drug dosage with a relatively low amount of nanotube vectors to save costs and to diminish the possible risk of agglomeration induced by high concentrations of nanotubes. The DOX loading efficiency was comparable to that found in other studies characterized by a high cargo loading capacity [12, 17]. In our research, the high loading amount of DOX is attributed to the large external surface area of O-LID-MWCNTs as well as the multiple bonding forms. Since the outer diameter of O-LID-MWCNTs was as much as 30–60 nm, the nanotubes possessed a large external surface area, which provided enough bonding sites for DOX. As described, there are two categories of DOX in this novel DDDSs. One is the external-surface-loaded DOX which is attached to the nanotubes through π-π stacking. Owing to the milder form of oxidation and the length selection procedure, this type of DOX accounts for most of the loading of DOX. The other category of DOX is species that serves as the tertiary blocker, which is bonded via hydrogen bonds and electrostatic interaction between FA and DOX. Notably, we deliberately decreased the initial added concentration of DOX and shortened the absorption time to avoid extremely high loading efficiency of the DOX ranging from 400 to 945% [30, 38], because the antitumor activity of DDDSs mainly comes from the synergistic effect afforded by different drugs, and it is unnecessary to load superabundant quantities of DOX from the practical and the economic points of view. Moreover, a huge adsorption capacity will lower the release rate of DOX under acidic conditions [17, 38], which would jeopardize the synergistic effect of DOX and CDDP and would, in turn, compromise the antitumor activity of the DDDS.
Remarkably, the loading efficiency of encapsulated CDDP of CDDP@O-LID-MWCNTs-PEG was significantly improved compared with previous investigations ranging from 3.38 to 62.1% [25, 26, 28], which we attribute to the huge inner space of O-LID-MWCNTs. However, as shown in Fig. 3, almost 48% of encapsulated CDDP leaves the PEGylated nanotubes within the first 6 h in PBS. This means that after the DOX loading procedure, the remaining CDDP in DDDSs only accounts for 52% of that in the CDDP@O-LID-MWCNTs-PEG. A solution to address this drug loss problem is urgently needed. As mentioned above, there is no potent interaction between encapsulated CDDP and the inner surface of O-LID-MWCNTs, and CDDP molecules are released from the nanotubes mainly through the diffusion mechanism originating from the concentration gradient between the release medium and the inner cavities of nanotubes [29]. To prevent the premature release of encapsulated CDDP during the DOX loading procedure, we dispersed CDDP powder into the DOX loading solution by sonication. As the concentration of CDDP increases in the PBS (release medium), the CDDP diffusion procedure was effectively inhibited. As a result, a high loading efficiency of 84.56% could still be obtained after DOX loading. Notably, although the excess CDDP powder was added into the DOX loading solution, the CDDP loading efficiency of the resulting DDDS was still a little bit lower than that of CDDP@O-LID-MWCNTs-PEG. The possible reason might be associated with the poor dispersibility of CDDP in PBS. During the stirring procedure, a minor amount of CDDP may re-precipitate and subsequently weaken the diffusion inhibition effect. Therefore, a small portion of CDDP leaves the nanotubes, resulting in a certain decrease in loading efficiency.
Ever since CNTs were discovered by Iijima in 1991, its potential carcinogenesis has attracted tremendous attention. Generally, genotoxicity and inflammation are regarded as critical events in carcinogenesis. Although the argue about whether CNTs are carcinogenic still exists, more available data favored the conclusion that CNTs in the form of short length, purification, functionalization and applied in low dose could exhibit relatively low or no carcinogenesis. CA Poland et al. reported that long MWCNTs could interact with mesothelium, leading to the inflammation and granulomas in mice, whereas short MWCNTs (<10 μm) did not have the interaction with mesothelium [39]. In a similar study, no sustained inflammatory reaction and carcinogenic response were detected after injecting short MWCNT into the rat peritoneal cavity [40]. As for our DDDS, owing to the fragile wall of LID-MWCNTs and effective oxidation procedure, the length of nanotube was significantly reduced to approximately 4 μm (tested by DLS) which was below the carcinogenesis-causing length. The influence of catalytic residues (Fe, Ni and V) on the toxicity of nanotubes is another issue [41]. Metal impurities existed in nanotubes may have contributed to carcinogenicity [42]. Based on our previous study, the pristine nanotubes did have an toxicity on cells [18]. Therefore, the highly purified nanotubes were obtained through the multi-step procedure prior to establishing DDDS. As shown in Fig. S2, the residual metal catalyst in purified nanotubes was significantly decreased (from 14.96 to 2.94%). And the purified nanotubes exerted no influence on the growth of L929 and MCF-7 cells (shown in Fig. S5). The functionalization is an important aspect to prevent aggregation and ensure a stable dispersion of nanotubes. With the high functionalization of nanotubes, the majority of nanotubes can be eliminated via urinary excretion [43, 44]. Evidence proved that the agglomeration state of nanotubes was closely related to the toxicity [45]. In addition, the functionalization can also decrease the toxicity of nanotubes and make them nonimmunogenic [46]. For our DDDS, the functionalization was conducted by grafting PEG to O-LID-MWCNTs. It has been demonstrated that PEGylated CNTs are able to extend the blood circulating time and reduce the uptake by the reticuloendothelial system [47]. The carcinogenesis of nanotubes could exhibit a dose dependent manner. Previous study shown that the number of subpleural fibrosis caused by nanotubes increased after 2 and 6 weeks after exposed to the high dose of nanotubes, however, none of this subpleural fibrosis could be observed after the low dose exposure [48]. Due to the high loading efficiency and the synergistic effect of both cargo drugs, the dose of nanotubes in our DDDS was relatively low (0.35–11 μg/ml) and its carcinogenesis was supposed to be low.
Besides, some studies reported that nanotubes can be enzymatic degraded. By incubating nanotubes in a cell-free system with horseradish peroxidase and low amounts of hydrogen peroxide, nanotubes was completely degraded [49]. The complete biodegradation of nanotubes was also achieved via neutrophil-derived human myeloperoxidase in another study. The degradation product did not elicit typical inflammatory and oxidative stress responses [50]. Hence, it is predictable that nanotubes which cannot be eliminated via urinary excretion might be somewhat degraded in vivo, and the degradation product would not be carcinogenetic.
In future studies, more efforts should be devoted to achieving active targeting property and to further improving the synergistic effect of DDDS. The active targeting effect of FA should be enhanced. The release rate of both drugs in acidic condition should be increased. Moreover, the optimum dose ratio of released CDDP and DOX should be achieved by tuning the dose of loaded DOX. Furthermore, near infrared irradiation could also be employed to promote drug release and to mediate tumor photothermal destruction. In this way, a novel comprehensive antitumor treatment that combines chemotherapy and photothermal therapy may be established.
5 Conclusions
In this study, a novel DDDS was established by employing highly purified and mildly oxidized LID-MWCNTs as vectors. With CDDP encapsulated inside the inner cavities of nanotubes and DOX attached to the external surfaces, the available space of the nanotube vectors was fully occupied, resulting in improved loading efficiencies for both drugs. Meanwhile, to achieve the pH-sensitive release profile of CDDP as well as to endow the DDDS with multifunctional properties, a three-level blocking strategy containing PEG, FA and DOX was applied to block the CDDP exits. Both cargo drugs in the novel DDDS exhibited pH-sensitive release profiles. Tested as an anticancer agent in MCF-7 cancer cell lines, the results verified the synergistically antitumor effect of the proposed DDDS. Furthermore, our results also provide the possibility for developing a novel comprehensive antitumor treatment that combines chemotherapy and photothermal therapy.
References
Parhi P, Mohanty C, Sahoo SK. Nanotechnology-based combinational drug delivery: an emerging approach for cancer therapy. Drug Discov Today. 2012;17(17-18):1044–52. doi:10.1016/j.drudis.2012.05.010
Sahoo SK, Parveen S, Panda JJ. The present and future of nanotechnology in human health care. Nanomedicine. 2007;3(1):20–31. doi:10.1016/j.nano.2006.11.008
Zhang X, Guo S, Fan R, Yu M, Li F, Zhu C, et al. Dual-functional liposome for tumor targeting and overcoming multidrug resistance in hepatocellular carcinoma cells. Biomaterials. 2012;33(29):7103–14. doi:10.1016/j.biomaterials.2012.06.048
Chun R, Kurzman ID, Couto CG, Klausner J, Henry C, MacEwen EG. Cisplatin and doxorubicin combination chemotherapy for the treatment of canine osteosarcoma: a pilot study. J Vet Intern Med. 2000;14(5):495–8. doi:10.1111/j.1939-1676.2000.tb02265.x
Guthrie TH Jr., Porubsky ES, Luxenberg MN, Shah KJ, Wurtz KL, Watson PR. Cisplatin-based chemotherapy in advanced basal and squamous cell carcinomas of the skin: results in 28 patients including 13 patients receiving multimodality therapy. J Clin Oncol. 1990;8(2):342–6.
Scambia G, DeVincenzo R, Ranelletti FO, Panici PB, Ferrandina G, Dagostino G, et al. Antiproliferative effect of silybin on gynaecological malignancies: synergism with cisplatin and doxorubicin. Eur J Cancer. 1996;32A(5):877–82. doi:10.1016/0959-8049(96)00011-1
Xu S-P, Sun G-P, Shen Y-X, Wei W, Peng W-R, Wang H. Antiproliferation and apoptosis induction of paeonol in HepG(2) cells. World J Gastroenterol. 2007;13(2):250–6.
Meng L, Zhang X, Lu Q, Fei Z, Dyson PJ. Single walled carbon nanotubes as drug delivery vehicles: targeting doxorubicin to tumors. Biomaterials. 2012;33(6):1689–98. doi:10.1016/j.biomaterials.2011.11.004
Du Z, Zhang S, Zhou C, Liu M, Li G. Dynamic layer-by-layer self-assembly of multi-walled carbon nanotubes on quartz wool for on-line separation of lysozyme in egg white. Talanta. 2012;94(0):104–10. doi:10.1016/j.talanta.2012.03.002
Ren Y, Pastorin G. Incorporation of hexamethylmelamine inside capped carbon nanotubes. Adv Mater.. 2008;20(11):2031–6. doi:10.1002/adma.200702292
Dhar S, Liu Z, Thomale Jr, Dai H, Lippard SJ. Targeted single-wall carbon nanotube-mediated Pt(IV) prodrug delivery using folate as a homing device. J Am Chem Soc. 2008;130(34):11467–76. doi:10.1021/ja803036e
Ren J, Shen S, Wang D, Xi Z, Guo L, Pang Z, et al. The targeted delivery of anticancer drugs to brain glioma by PEGylated oxidized multi-walled carbon nanotubes modified with angiopep-2. Biomaterials. 2012;33(11):3324–33. doi:10.1016/j.biomaterials.2012.01.025
Gerweck LE, Seetharaman K. Cellular pH gradient in tumor versus normal tissue: potential exploitation for the treatment of cancer. Cancer Res. 1996;56(6):1194
Wike JL. The relevance of tumour pH to the treatment of malignant disease. Radiother Oncol. 1984;2(4):343
Du C, Deng D, Shan L, Wan S, Cao J, Tian J, et al. A pH-sensitive doxorubicin prodrug based on folate-conjugated BSA for tumor-targeted drug delivery. Biomaterials. 2013;34(12):3087–97. doi:10.1016/j.biomaterials.2013.01.041
Yin Q, Shen J, Zhang Z, Yu H, Li Y. Reversal of multidrug resistance by stimuli-responsive drug delivery systems for therapy of tumor. Adv Drug Delivery Rev. 2013;65(13-14):1699–715. doi:10.1016/j.addr.2013.04.011
Zhang X, Meng L, Lu Q, Fei Z, Dyson PJ. Targeted delivery and controlled release of doxorubicin to cancer cells using modified single wall carbon nanotubes. Biomaterials. 2009;30(30):6041–7. doi:10.1016/j.biomaterials.2009.07.025
Sui L, Yang T, Gao P, Meng A, Wang P, Wu Z, et al. Incorporation of cisplatin into PEG-wrapped ultrapurified large-inner-diameter MWCNTs for enhanced loading efficiency and release profile. Int J Pharm. 2014;471(1–2):157–65. doi:10.1016/j.ijpharm.2014.05.022
Liu J, Zhong L, Zhang J, Luo T, Zhou J, Zhao X, et al. Hollow mesoporous silica nanoparticles facilitated drug delivery via cascade pH stimuli in tumor microenvironment for tumor therapy. Biomaterials. 2016;83:51–65.
Ma G, Zhang C, Zhang L, Sun H, Song C, Wang C, et al. Doxorubicin-loaded micelles based on multiarm star-shaped PLGA–PEG block copolymers: influence of arm numbers on drug delivery. J Mater Sci: Mater Med. 2015;27(1):17 doi:10.1007/s10856-015-5610-4
Chen Q. Functional nanomaterials of NaYF4: Yb/Er- MWCNT: synthesis, characterization and applications in targeted diagnosis and therapy of tumor cell. Scientia Sinica Chimica. 2011;41(5):798 doi:10.1360/032010-860
Narayanan NK, Nargi D, Randolph C, Narayanan BA. Liposome encapsulation of curcumin and resveratrol in combination reduces prostate cancer incidence in PTEN knockout mice. Int J Cancer. 2009;125(1):1–8. doi:10.1002/ijc.24336
Tekade RK, Dutta T, Gajbhiye V, Jain NK. Exploring dendrimer towards dual drug delivery: pH responsive simultaneous drug-release kinetics. J Microencapsul. 2009;26(4):287–96. doi:10.1080/02652040802312572
Katragadda U, Teng Q, Rayaprolu BM, Chandran T, Tan C. Multi-drug delivery to tumor cells via micellar nanocarriers. Int J Pharm. 2011;419(1-2):281–6. doi:10.1016/j.ijpharm.2011.07.033
Li J, Yap SQ, Yoong SL, Nayak TR, Chandra GW, Ang WH, et al. Carbon nanotube bottles for incorporation, release and enhanced cytotoxic effect of cisplatin. Carbon. 2012;50(4):1625–34. doi:10.1016/j.carbon.2011.11.043
Tripisciano C, Kraemer K, Taylor A, Borowiak-Palen E. Single-wall carbon nanotubes based anticancer drug delivery system. Chem Phys Lett. 2009;478(4-6):200–5. doi:10.1016/j.cplett.2009.07.071
Wang J. Effects of ultrasonic radiation intensity on the oxidation of single-walled carbon nanotubes in a mixture of sulfuric and nitric acids. Nano. 2013;8(4):46–54.
Guven A, Rusakova IA, Lewis MT, Wilson LJ. Cisplatin@US-tube carbon nanocapsules for enhanced chemotherapeutic delivery. Biomaterials. 2012;33(5):1455–61. doi:10.1016/j.biomaterials.2011.10.060
Li J, Yap SQ, Chin CF, Tian Q, Yoong SL, Pastorin G, et al. Platinum(IV) prodrugs entrapped within multiwalled carbon nanotubes: selective release by chemical reduction and hydrophobicity reversal. Chem Sci. 2012;3(6):2083–7. doi:10.1039/c2sc01086k
Liu Z, Sun X, Nakayama-Ratchford N, Dai H. Supramolecular chemistry on water-soluble carbon nanotubes for drug loading and delivery. ACS Nano. 2007;1(1):50–56. doi:10.1021/nn700040t
Niu L, Meng L, Lu Q. Folate-conjugated PEG on single walled carbon nanotubes for targeting delivery of doxorubicin to cancer cells. Macromol Biosci. 2013;13(6):735–44. doi:10.1002/mabi.201200475
Huang H, Yuan Q, Shah JS, Misra RD. A new family of folate-decorated and carbon nanotube-mediated drug delivery system: synthesis and drug delivery response. Adv Drug Deliv Rev. 2011;63(14-15):1332–9. doi:10.1016/j.addr.2011.04.001
Cheng F-F, Zhang J-J, Xu F, Hu L-H, Abdel-Halim ES, Zhu J-J. pH-Sensitive polydopamine nanocapsules for cell imaging and drug delivery based on folate receptor targeting. J Biomed Nanotechnol. 2013;9(7):1155–63. doi:10.1166/jbn.2013.1611
Yi P, Chen KL. Influence of surface oxidation on the aggregation and deposition kinetics of multiwalled carbon nanotubes in monovalent and divalent electrolytes. Langmuir. 2011;27(7):3588–99. doi:10.1021/la104682b
Saleh NB, Pfefferle LD, Elimelech M. Aggregation kinetics of multiwalled carbon nanotubes in aquatic systems: measurements and environmental implications. Environ Sci Technol. 2008;42(21):7963–9. doi:10.1021/es801251c
Ross JF, Chaudhuri PK, Ratnam M. Differential regulation of folate receptor isoforms in normal and malignant tissues in vivo and in established cell lines. Physiologic and clinical implications. Cancer. 1994;73(9):2432
Weitman SD, Lark RH, Coney LR, Fort DW, Frasca V, Zurawski V Jr, et al. Distribution of the folate receptor GP38 in normal and malignant cell lines and tissues. Cancer Res. 1992;52(12):3396
Wang Y, Yang S-T, Wang Y, Liu Y, Wang H. Adsorption and desorption of doxorubicin on oxidized carbon nanotubes. Colloids Surf B. 2012;97:62–69. doi:10.1016/j.colsurfb.2012.04.013
Poland CA, Duffin R, Kinloch I, Maynard A, Wallace WAH, Seaton A, et al. Carbon nanotubes introduced into the abdominal cavity of mice show asbestos-like pathogenicity in a pilot study. Nat Nanotechnol. 2008;3(7):423–8. doi:10.1038/nnano.2008.111
Muller J, Delos M, Panin N, Rabolli V, Huaux F, Lison D. Absence of carcinogenic response to multiwall carbon nanotubes in a 2-year bioassay in the peritoneal cavity of the rat. Toxicol Sci. 2009;110(2):442–8.
Grubek-Jaworska H, Nejman P, Czumińska K, Przybyłowski T, Huczko A, Lange H, et al. Preliminary results on the pathogenic effects of intratracheal exposure to one-dimensional nanocarbons. Carbon. 2006;44(6):1057–63.
Takagi A, Hirose A, Nishimura T, Fukumori N, Ogata A, Ohashi N, et al. Induction of mesothelioma in p53 mouse by intraperitoneal application of multi-wall carbon nanotube. J Toxicol Sci. 2008;33:105–16.
Bianco A, Kostarelos K, Prato M. Making carbon nanotubes biocompatible and biodegradable. Chem Commun. 2011;47(37):10182–8.
Lacerda L, Ali-Boucettal H, Herrero MA, Pastorin G, Bianco A, Prato M, et al. Tissue histology and physiology following intravenous administration of different types of functionalized multiwalled carbon nanotubes. Nanomedicine. 2008;3(2):149–61. doi:10.2217/17435889.3.2.149
Fraczek A, Menaszek E, Paluszkiewicz C, Blazewicz M. Comparative in vivo biocompatibility study of single- and multi-wall carbon nanotubes. Acta Biomater. 2008;4(6):1593–602.
Gulati N, Gupta H. Two faces of carbon nanotube: toxicities and pharmaceutical applications. Crit Rev Ther Drug Carrier Syst. 2012;29(1):65–88.
Liu Z, Chen K, Davis C, Sherlock S, Cao Q, Chen X, et al. Drug delivery with carbon nanotubes for in vivo cancer treatment. Cancer Res. 2008;68(16):6652–60.
Ryman-Rasmussen JP, Cesta MF, Brody AR, Shipley-Phillips JK, Everitt JI, Tewksbury EW, et al. Inhaled carbon nanotubes reach the subpleural tissue in mice. Nat Nanotechnol. 2009;4(11):747–51.
Allen BL, Kichambare PD, Gou P, Vlasova II, Kapralov AA, Konduru N, et al. Biodegradation of single-walled carbon nanotubes through enzymatic catalysis. Nano Lett. 2008;8(11):3899–903.
Kagan VE, Konduru NV, Feng W, Allen BL, Conroy J, Volkov Y, et al. Carbon nanotubes degraded by neutrophil myeloperoxidase induce less pulmonary inflammation. Nat Nanotechnol. 2010;5(5):354–9.
Acknowledgements
We would like to thank Pro Yunmao Liao and Dr. Xibo Pei for their constructive discussion. This work was supported by Tianjin Natural Science Foundation (grant no. 16JCZDJC32800) and Sichuan Natural Science Foundation (grant no. 2014SZ0201).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Tao Yang and Zhenzhen Wu contributed equally to this work.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Yang, T., Wu, Z., Wang, P. et al. A large-inner-diameter multi-walled carbon nanotube-based dual-drug delivery system with pH-sensitive release properties. J Mater Sci: Mater Med 28, 110 (2017). https://doi.org/10.1007/s10856-017-5920-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10856-017-5920-9