
Abstract
Carbon-bonded alumina (Al2O3–C) refractories were fabricated using unmodified (3–1, 1–0, and 0.045 mm) and Si powder-modified tabular alumina aggregates (3–1 mm) as raw materials, in addition to Si powder, ultra-fine α–Al2O3 powder, and carbon black as fines, and a thermosetting phenolic resin as the binder. The microstructures and phase compositions of the samples were analyzed by scanning electron microscopy and X-ray diffraction. After the mixing and wearing test, the Si powder-modified tabular alumina aggregates exhibited a low Si powder dropping ratio, thereby meeting the requirements to prepare Al2O3–C refractories. The addition of Si powder-modified tabular alumina aggregates to the material had little effect on the thermal shock resistance of the Al2O3–C refractory and was beneficial to improving the cold modulus of rupture of the material. Since the surface of the added modified aggregate was not dense, diffusion and mass transfer were promoted during the oxidation process, thereby leading to a slight decrease in the oxidation resistance of the material. The introduction of Si powder-modified tabular alumina aggregates increased the amount of Si powder particles surrounding the aggregates, resulting in the in situ generation of additional SiC fibers during high temperature heat treatment. Ultimately, this strengthened the interfacial bonding between the matrix and the aggregates. In addition, compared with the hot modulus of rupture (HMOR) of sample S0 (containing 50 wt.% tabular alumina aggregates), the HMOR of sample S2 (containing 51 wt.% modified tabular alumina aggregates with 1 wt.% Si powder coating) was 30% higher.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Slide gate plates are commonly used as functional refractory components for steelmaking and continuous casting by regulating the flow of molten steel in the ladle and tundish. These components are typically subjected to extreme conditions, such as slag erosion, severe thermal shock, chemical attack, and oxidation, and are therefore required to meet strict standards in terms of their high temperature performances, including a high temperature strength and resistance to thermal shock, corrosion, and oxidation. Slide plate refractory materials are typically composed of oxide, oxide–carbon, and oxide-non-oxide compounds [1,2]. At present, Al2O3–C and Al2O3–ZrO2–C are widely used for manufacturing slide gate plate refractories, due to their relatively advantageous high temperature performances and their adaptability [3,4]. Through the systematic analysis of the wear behaviors of these components, the sliding surface damage, bore enlargement, and crack propagation have been highlighted as the primary causes of component degradation. Numerous statistics have illustrated that the sliding surface damage accounts for > 50% of wear incidents and therefore constitutes the main source of sliding plate damage [4]. High temperature friction can cause the hot peeling of aggregates from the matrix due to the weak interfacial bonding in the slide plate and so can damage the sliding surface. One of the main research directions for alleviating the complication of sliding surface wear is therefore focused on improving the bonding strength between the matrix and the aggregates.
Numerous researchers have explored extensive modification techniques for application to raw materials by investigating the relationship between the aggregates and the matrix, and as a result, they have been able to regulate the raw material composition and distribution, along with the in situ formation of the non-oxide reinforcing phase on the aggregate surface [5]. These efforts have successfully enhanced the bonding strength between the matrix and the aggregates and improved the high temperature mechanical performances of the materials. Further, Wang et al. [6] and Behera et al. [7] investigated the effects of expanded graphite addition in alumina carbon materials and successfully improved their thermal shock resistance properties. Furthermore, Si-hybrid-expanded graphite (β-SiC and SiOx balls) exhibited an even better effect. In addition, Liao et al. [8] studied the enhancement effects of nano-carbon black and multi-walled carbon nanotubes on low-carbon alumina carbon refractories, revealing the toughening effects of the in situ formed multi-walled carbon nanotubes and SiC species on the latter. Furthermore, Wan et al. [9] employed corundum aggregates as raw materials to prepare lightweight refractories through an MgO carbothermal reduction method, which enhanced the bonding strength between the corundum aggregates and the spinel matrix, thereby improving the high temperature performance of the spinel corundum refractories. Moreover, Wang et al. [10] used high-activity metallurgical coke, rather than graphite, as a raw material to accelerate the formation of β–SiC whisker phases, and successfully formed an interconnected structure within the material, which strengthened the interfacial bonding. However, the works discussed above primarily focused on the modification of the raw materials (e.g., the carbon sources and binders), thereby leaving much room for exploring surface modification techniques for the larger bulk aggregates.
During the manufacturing process of high temperature-sintered Al2O3–C and Al2O3–ZrO2–C sliding plates, a certain amount (2–4 wt.%) of Si powder is often added. On the one hand, Si powder is introduced into the system as an antioxidant, which reacts with CO to form SiO2 species that fill the pores or cover the surface of the material, thereby preventing oxygen diffusion into the interior. On the other hand, non-oxide reinforcing phases, such as SiC, Si3N4, and SiALON [8,11,12,13,14,15], are formed when Si powder reacts with the C and N2 species in the material at high temperatures, effectively improving the high temperature mechanical properties of the material in the process. In our previous research [16], we successfully prepared Al powder-coated modified tabular alumina aggregates and introduced them into Al2O3–Al–C sliding plates to form a "chain-like ball structure" at high temperatures. This unique structure strengthened the interfacial bonding between the matrix and the aggregates, substantially improving the thermal shock resistance and hot modulus of rupture (HMOR) of the material. Hence, we theorized that the coating of Si powder-modified tabular corundum aggregates followed by their introduction into Al2O3–C sliding plates could prove to be an effective method for enhancing the overall high temperature performance of the material. The SiC species that forms in situ on the surface of the aggregates during heating increases the gripping force of the matrix, effectively preventing the peeling off of aggregates from the material and improving the wear resistance of the sliding surface while prolonging the service life of the refractory component. Herein, we investigate the effects of Si powder-modified tabular alumina aggregates on the phase compositions, microstructures, and properties of Al2O3–C materials.
Experimental
Raw materials
Tabular alumina (Al2O3 > 98.5%, Fe2O3 < 0.3%, SiO2 < 0.2%; 3–1 mm, 1–0 mm, ≤ 0.045 mm, Shandong Wanbang New Material Co., Ltd., China), ultra-fine α-Al2O3 powder (Al2O3 > 99.5%, d50 = 2 μm, Zhengzhou Yufa Fine Porcelain Technology Co., Ltd., China), Si powder (Si > 99.0%, ≤ 0.045 mm, Anyang Oriental Metallurgy Co., Ltd., China), and carbon black (N220: 25–20 nm, ≥ 99.0% fixed carbon; Wuhan Kebang New Material Co., Ltd., China) were used as raw materials in the preparation of the Al2O3-C samples. Thermoset phenolic resin (viscosity = 10 Pa·S, > 45% fixed carbon; Zhengzhou Tiancheng Abrasive Material Co., Ltd., China) was used as the binder. Glycol (≥ 99.5%; Tianjin Fuyu Fine Chemical Co., Ltd, China) was used as the dilution solvent.
Preparation of modified tabular alumina aggregates
Initially, the thermoset phenolic resin and the glycol were manually premixed in a 1:1 weight ratio using a glass rod, and this mixture was used as the binder for the following experimental procedure. Tabular alumina (3–1 mm) was used as the starting material for modification. Thus, the tabular alumina aggregates were added to a mixer (NRJ-411A, Wuxi Jianyi Instrument & Machinery Co., Ltd., China) and blended for 2 min, after which time the resin–glycol binder was added, and blending was continued for 3 min. Subsequently, 2 or 4 wt.% Si powder (relative to the aggregates) was introduced into the fully wetted aggregates and stirring continued for a further 5 min until the surfaces of the alumina aggregates were uniformly coated by the binder. Finally, the mixture was removed from the mixer and allowed to stand under an air atmosphere for 4 h prior to curing at 180 °C over 12 h in an oven (GZX-FX202-1, Shanghai Shishu Instrument Co., Ltd., China) to give the Si powder-modified tabular alumina aggregates. It should be noted here that due to the high viscosity of the phenolic resin, the modified aggregates tend to agglomerate and form blocks during the curing process. Therefore, the modified aggregates must be separated manually after curing. According to the different coating amounts of Si powder (i.e., 2 and 4 wt.%), the modified tabular alumina aggregates (3–1 mm) were marked as M-S2 and M-S4, respectively.
Τhe bonding strength of the Si powder on the surface of the modified tabular alumina was closely monitored in the subsequent experimental process, as the action of various external forces (e.g., friction, extrusion, and kneading) can potentially cause the Si powder to detach during preparation of the refractory samples. The coating strength between the Si powder and the tabular alumina was determined through mixing and wear experiments. Initially, the modified tabular alumina aggregates (750 g) were placed into the mixer and rotated at 120 rpm for 10 min. Subsequently, the aggregates were sieved using a 0.9-mm sieve. The sieved aggregates were then placed into the mixer and rotated for 10 min prior to passing through the sieve once again. After three cycles, the Si powder drop ratio was characterized by calculating the weight of dropped powder compared to the total weight of coated powder.
Al2O3-C sample preparation
Table 1 summarizes the formulation of the samples used in this work. More specifically, S0, containing 50 wt.% tabular alumina aggregates (3–1 mm), was prepared and used as a reference sample. The percentage of Si powder-modified alumina aggregates (M-S2, M-S4) contained in samples S2 and S4 were 51 and 52 wt.%, respectively; the Si powder content of the samples was reduced to 2 wt.% and 1 wt.%, respectively. All powders (tabular alumina fine, ultra-fine α-Al2O3, carbon black, and Si) were precisely measured and premixed for 5 min by planetary ball-milling (YXQM-4L, MITR Co., Ltd., China). Following the addition of the alumina aggregates to the mixer, the thermoset phenolic resin (3 wt.%) was introduced to wet their surfaces, and mixing was continued for a further 3 min. After slowly adding all premixed powders into the mixer, mixing was continued for ~ 15 min. The resulting mixture was then discharged, and the mixture was aged in an airtight bag for 4 h. After this time, the mixture was homogeneously pressed for 10 s under standard pressure conditions (150 MPa), to form 25 × 25 × 150 mm samples. All samples were held under air at 25 °C for 24 h and then subjected to the following curing process: 60 °C for 6 h, 110 °C for 6 h, and 180 °C for 12 h. Finally, the desired samples were successfully obtained after air cooling.
The apparent porosity and bulk density of the prepared samples were then examined according to the Chinese standard (GB/T 2997–2015 [17]), based on the Archimedes principle. The cold modulus of rupture (CMOR) of each sample was determined in accordance with the Chinese standard (GB/T 3001–2017 [18]), while the HMOR was examined according to the GB/T 3002–2017 [19] standard under a carbon-buried reducing atmosphere at 1400 °C for 0.5 h. To investigate the thermal shock resistance, each sample was placed in a carbon bed and fired at 1400 °C for 3 h. After cooling under air, the samples were separated in two groups. In the first group, the CMOR was determined using a computer-controlled pressure testing machine (WHY-300/10, Shanghai Hualong Testing Instrument Co., Ltd, China) and the force–displacement curve was recorded simultaneously. In the second group, the samples were placed in an electrical furnace under ambient atmosphere, then heated at 1100 °C for 30 min, and subsequently cooled under air for 30 min. Subsequently, the cooled samples were reheated at 1100 °C for 30 min under air and then air-cooled once again for 30 min. After three cycles, the thermal shock resistance was investigated based on the CMOR values before and after thermal shock of the samples. Furthermore, the oxidation resistance of the samples was characterized based on the average oxidation layer thicknesses. More specifically, after the identification of two spots in each white (oxidized) section of the sample cross section following thermal shock, the thickness was measured, and the average oxidation layer thickness was determined by calculating an average of eight values. Moreover, material was taken from the inner part of the samples that had been treated at 1400 °C, where the black color was maintained. This material was ground into a powder (≤ 0.04 mm), and the phase composition was determined by means of X-ray diffraction (XRD, D8 Advance, Bruker, Germany) using Cu Kα1 radiation over the 2θ diffraction range of 10–80° with a step size of 0.02°). The polished surface and fracture morphology of the samples after heat treatment at 180 °C and 1400 °C, respectively, were investigated using scanning electron microscopy (SEM, INSPECT F50, FEI, USA).
Results and discussion
Characterization of modified tabular alumina aggregates
The Si powder drop ratio of the samples was measured after performing the mixing and wear experiments on the modified tabular alumina, as illustrated in Fig. 1. During the mixing and wear experiments, the Si powder particles attached on the rough surface of the inner coating layer sustained larger a stress and began to gradually detach (superficial Si powder drop). The Si powder drop ratios of the M-S2 and M-S4 samples gradually increased with the extension of the mixing and wear time. Although the measured drop ratio of M-S2 was slightly higher than that of M-S4, it remained within an acceptable range, which met the requirements for the follow-up experiment.

Si powder drop ratio after mixing and wear experiments
The morphologies of the tabular alumina aggregates before and after modification are shown in Fig. 2. It was observed that the surface of the unmodified aggregates was white and that of the modified aggregates was gray. With an increase in the coating amount of Si powder, the color of the modified aggregate surface gradually darkened. The microstructures of the Si powder particles and samples (after thermally treating at 180 °C) are illustrated in Fig. 3. The Si powder particles (Fig. 3a) show a highly heterogeneous size distribution, with many particles falling within the 0–20 μm range, and quite a few exceeding 30 μm. The bright particles observed in Fig. 3b are Si powder particles that are distributed randomly within the matrix. In contrast, Si powder was barely observed in the area surrounding the unmodified aggregates due to the small amount of Si powder employed; however, the coating layer was clearly identifiable around the modified aggregate. More specifically, a thin, discontinuous Si powder coating layer is observed around the tabular alumina aggregates in Fig. 3c, whereas a clearer coating layer is observed in Fig. 3d.

Morphology of unmodified and modified tabular alumina aggregates (treated at 180 °C)

SEM micrograph of Si powder particles a and Al2O3–C samples thermally treated at 180 °C for 12 h: S0 sample (b) and S2 sample (c and d)
Effects of modified tabular alumina aggregates on material properties
Physical properties
The apparent porosity and bulk density of the S0, S2, and S4 samples are reported in Fig. 4. As shown, the bulk density demonstrated an initially increasing and then decreasing trend, which was in complete contrast to the apparent porosity. The bulk density of the samples reached its maximum with a 2 wt.% Si powder coating, thereby implying that the rough surfaces and pores of the tabular alumina aggregates had been optimally filled up by Si powder particles to enhance the densification of the specimen. However, as the amount of coated Si powder began to increase further, the bulk density of the samples decreased.

Apparent porosity and bulk density of the Al2O3–C samples thermally treated at 180 °C for 12 h
Thermo-mechanical properties
The HMOR results for the S0, S2, and S4 samples are reported in Fig. 5. More specifically, the HMOR of samples S2 and S4, which contained added modified tabular alumina aggregates, were higher than that of sample S0. The total amount of Si powder added to all samples was 3 wt.%, but the addition methods were different. For example, 0, 1, and 2 wt.% of Si powder were applied in sequence to coat the surfaces of the aggregates, while the remaining Si powder (3, 2, and 1 wt.%, respectively) was directly introduced into the material. The sample coated with 1 wt.% Si powder exhibited the highest HMOR value, which was approximately 30% higher than that of the reference sample. However, the HMOR value of the 2 wt.% Si powder coated sample was not significantly improved.

HMOR values of the three samples tested under a reducing atmosphere (1400 °C)
The CMOR values of the three samples before and after thermal shock are reported in Fig. 6. Compared with sample S0, the strength retention rates of the S2 and S4 samples were slightly lower; however, they still reached > 95%. Moreover, the CMOR values of the samples bearing modified aggregates were significantly higher than those of the S0 before and after the thermal shock tests. This result indicates that the addition of modified aggregates has little effect on the thermal shock resistance of the sample and that it was beneficial in terms of improving the CMOR of the sample.

CMOR values before and after thermal shocks of Al2O3–C samples
The oxidation layers of the samples after thermal shock testing are illustrated in Table 2. From a cross-sectional viewpoint, the white part surrounding the samples corresponded to the oxidation layer, which was loose compared with the black area in the middle. After adding the modified aggregates, the oxide layer thickness of the samples increased slightly. Moreover, compared with the S2 sample, the thickness of the oxide layer of sample S4 increased to a lesser extent.
Phase composition and microstructure
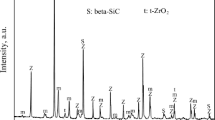
Figure 7 illustrates the XRD patterns of the samples that were thermally treated for 3 h at 1400 °C in the carbon bed. As can be observed, the crystalline phases of the samples primarily comprised α–Al2O3 and β-SiC. Interestingly, most of the Si species present in the S0 sample reacted with C and CO to form SiC, although weak Si peaks were detected, thereby indicating that there was still a small amount of residual Si in this sample.

XRD patterns of the Al2O3–C samples thermally treated at 1400 °C × 3 h
The microstructures of samples S0 and S2, which were also fired at 1400 °C for 3 h, are illustrated in Fig. 8. As can be seen, both samples exhibited needle-like and fiber cluster-like non-oxide SiC species on the surfaces of their aggregates. In the case of S0 (Fig. 8a, b, and c), the amount of non-oxide SiC species was lower, and this was mainly located on the smooth surfaces of the aggregates. Compared with S0, a larger amount of non-oxide SiC species formed between the aggregates and the matrix in sample S2 (Fig. 8d, e and f), which were tightly wrapped around the aggregates, and induced a bridge-like effect. In addition, well-developed non-oxide crystalline phase was found to form in sample S2, along with clear contours.

SEM micrographs of the Al2O3–C samples (fired at 1400 °C): S0 (a, b and c) and S2 (d, e and f)
Discussion
The intrinsic properties and microstructural evolution of each non-oxide phase are elucidated in Fig. 9. When an appropriate amount of Si powder covers the tabular alumina surfaces, the surfaces of the aggregates become smooth and the bulk density increases (Fig. 4). Furthermore, the distribution of Si powder in the material is effectively regulated, thereby facilitating the accumulation of Si powder at the aggregates–matrix interface. At elevated temperatures, the Si powder reacted with C and CO in the material to produce fibrous, needle-like, and non-oxide SiC phases, generating a "chain-like ball structure" with bridge-like features in the process, which strengthened the interface between the aggregates and the matrix. Consequently, the binding force of the matrix on the aggregates was substantially augmented, thereby improving the high temperature performance of the material.

Schematic illustration of Si powder-modified tubular alumina aggregates
At higher temperatures, the microstructure of sample S2 presented significantly higher amounts of fibrous SiC phases compared with sample S0 (Fig. 8). This phenomenon further verified that coating the surface of tabular alumina with Si powder benefitted the formation of non-oxide strengthening phases, which enhanced the HMOR of the material. This was also confirmed by the HMOR of S2, which was 30% higher than that of S0 (Fig. 5). Since the total amount of Si powder added to the samples was 3 wt.%, the content of Si powder in the matrix of sample S4 was only 1 wt.%. Therefore, the amount of SiC generated in the matrix of sample S4 was lower, which ultimately affected the strength of the material. In contrast, when 2 wt.% of Si powder coating was employed, the Si powder layer coated on the surface of the modified aggregates was thicker, which affected the bonding between the aggregates and the matrix. Moreover, with a Si powder coating of 2 wt.%, the HMOR of sample S4 was lower than that of sample S2, and so it was inferred that the optimal coating amount of the Si powder was ~ 1 wt.%.
Due to the direct introduction of the fine Si powder into the matrix in sample S0, the Si powder may be inhomogeneously distributed within the material. Since the Si powder-coated modified aggregates used phenolic resin as the binder, fixed carbon was formed upon heating to coat the Si powder on the surfaces of the modified aggregates. This coating layer is not dense, and thermal decomposition of the phenolic resin therefore causes gap formation on the particle surfaces, which is conducive to gas diffusion and mass transfer. Compared with the Si powder coated on the modified aggregates, the Si powder distributed in the matrix may have a reduced chance of coming into contact or reacting with C, CO, and N2, thereby lowering the reaction rate of the Si powder during heating at 1400 °C for 3 h. As a result, additional residual Si particles were present in the S0 sample compared with samples S2 and S4 (Fig. 7). In the thermal shock resistance experiment, the CMOR of sample S0 increased slightly (Fig. 6), which was due to the continued reaction of residual Si, and this ultimately played a role in strengthening the material.
Limited by the modification process, the coating layer on the surface of the modified aggregates was not dense, and the thermal decomposition of the coating binder resulted in gaps on the surfaces of the modified aggregates. Both factors are conducive to promoting diffusion and mass transfer during the oxidation process and therefore lead to a slight increase in the thickness of the oxide layer. Following the heat treatment at 1400 °C, a large number of SiC whiskers were formed on the surfaces of the modified aggregates. These SiC whiskers are easily oxidized to SiO2, and the volume expansion produced during this process hinders the progress of the oxidation reaction. Since the surface of the modified aggregate in sample S4 produced a greater quantity of SiO2 than sample S2, the thickness of the oxide layer of sample S4 increased to a lesser extent.
The chemical reactions that occurred at high temperatures between the Si particles coated on the alumina aggregate surfaces and the C/CO species in the material, through which the non-oxide SiC phases were formed, are elucidated in chemical Eqs. (1–7) [20,21,22,23,24], in which Eqs. (1–4) are the basic reaction reactions, and Eqs. (5–7) were obtained after determining reactions (1–4).
Based on our results, it was apparent that the thermal condition of the formation of the non-oxide phase was sufficient due to the fact that the material system was carbon-containing. At 1400 °C, the Gibbs free energies (ΔrGθ) of Eqs. (4–7) were determined to be − 51.80, − 67.27, 174.24, and 68.95 kJ/mol, respectively. These results indicate that the predominant reactions taking place during the formation of SiC are those of Eqs. (4 and 5), and the thermodynamic probability of reaction (5) taking place is larger than that of (4). In addition, from a kinetics perspective, the diffusion mass transfer was enhanced after the oxidation of Si(s) to SiO(g), which in turn accelerated the SiC formation reaction.
ΔGθ= – 231752 + 179.74 T
Since the surface density of the Si powder-coated modified aggregates was lower than that of the unmodified aggregates (Fig. 2), it was considered that a greater number of gaps must be present between the modified aggregate and the matrix; this was conducive to the diffusion and mass transfer of SiO(g) and promoted the formation of SiC. Therefore, the amounts of fibrous and needle-like SiC observed around the tabular alumina aggregates in Figs. 8e and f were greater than those observed in Figs. 8b and c. In addition, it should be pointed out here the mass transfer diffusion in the gas phase contributed to the production of fibrous SiC, which had a positive effect on the HMOR of the material. Due to the fact that a greater amount of fibrous SiC was generated around the modified aggregates, sample S2 exhibited a higher HMOR.
Overall, it was demonstrated that the incorporation of Si powder-modified tabular alumina aggregates into Al2O3–C materials regulated the distribution of Si powder within the aggregates, and as a result, part of the Si powder was distributed on the aggregate surfaces instead of being incorporated into the matrix. In addition, the fibrous, needle-like non-oxide SiC phases strengthened the interfacial bonding between the modified aggregates and the matrix, owing to their bridge-like structures. As a result, the thermo-mechanical properties (e.g., the HMOR) were improved and the service life of the sliding refractory plate was prolonged.
Conclusions
Si powder-modified tabular alumina aggregates bearing high-strength coatings were prepared, and these aggregates were tested and verified to meet the requirements for preparing reliable carbon-bonded alumina (Al2O3–C) refractories. Initially, the tabular alumina aggregate surfaces were coated with the desired quantity of Si powder (1 wt.%), and this was conducive to the in situ high temperature formation of fibrous, needle-like SiC phases with bridge-like structures. Thus, the bonding strength between the aggregates and matrix was enhanced at the interface, which improved both the density and the hot modulus of rupture of the material. Following the addition of Si powder-modified tabular alumina aggregates, the material maintained a high cold modulus of rupture before and after the thermal shock tests. Although the thermal shock resistance of the samples containing the modified aggregates was slightly reduced, the residual strength ratio remained above 95%. Moreover, since the surfaces of the modified aggregates were not dense, diffusion and mass transfer were promoted during the oxidation process. The addition of these modified aggregates therefore caused a slight decrease in the oxidation resistance of the material. This study proposes a novel method for the fabrication of modified tabular corundum aggregates, which has important potential applications in steelmaking and metallurgy.
References
Zhong XC (2008) Progress in research and development of refractory oxide-nonoxide composites. China’s Refract 17:1–5. https://doi.org/10.3969/j.issn.1004-4493.2008.02.001
Shi K, Xia Y (2018) Evolution of material systems of slide gates for steelmaking. Refractories 52:230–236. https://doi.org/10.3969/j.issn.1001-1935.2018.03.019
Ban JJ, Zhou CJ, Feng L, Jia QL, Liu XH, Hu JH (2020) Preparation and application of ZrB2–SiCw composite powder for corrosion resistance improvement in Al2O3–ZrO2–C slide plate materials. Ceram Int 46:9817–9825. https://doi.org/10.1016/j.ceramint.2019.12.255
Shi K, Zhong XC (2007) Properties and application of Al–Si metal bonded Al2O3–C slide plate. Refractories 41:205–207. https://doi.org/10.3969/j.issn.1001-1935.2007.03.012
Gu Q, Ma T, Zhao F, Jia QL, Liu XX, Liu GQ, Li HX (2020) Enhancement of the thermal shock resistance of MgO–C slide plate materials with the addition of Nano–ZrO2 modified magnesia aggregates. J Alloys Compd 847:156339. https://doi.org/10.1016/j.jallcom.2020.156339
Wang QG, Li YW, Sang SB, Jin SL (2018) Enhanced mechanical properties of Al2O3–C refractories with silicon hybridized expanded graphite. Mater Sci Eng A 709:160–171. https://doi.org/10.1016/j.msea.2017.10.046
Behera SK, Mishra B (2015) Strengthening of Al2O3–C slide gate plate refractories with expanded graphite. Ceram Int A 41:4254–4259. https://doi.org/10.1016/j.ceramint.2014.11.092
Liao N, Li YW, Sang SB (2017) Effects of silicon and microsilica additive on microstructure and mechanical properties of Al2O3–C multi-walled carbon nanotubes refractories. J Chinese Ceram Soc 45:433–440
Wan QF, Yin HF, Tang Y, Yuan HD, Ren XH, Gao K, Xin YL, Liu YC (2015) Effect of aggregate on aggregate/spinel matrix bonding interface and mechanical performance of lightweight spinel-bonded refractory. Ceram Int A 46:18362–18365. https://doi.org/10.1016/j.ceramint.2020.04.095
Wang HF, Bi YB, Zhou NS, Zhang HJ (2016) Preparation and strength of SiC refractories with in situ beta-SiC whiskers as bonding phase. Ceram Int A 42:727–733. https://doi.org/10.1016/j.ceramint.2015.08.172
Zhu BQ, Zhu YN, Lin XC, Zhao F (2013) Effect of ceramic bonding phases on the thermo-mechanical properties of Al2O3–C refractories. Ceram Int 39:6069–6076. https://doi.org/10.1016/j.ceramint.2013.01.024
Chen XY, Zhang Q, Zhou Y, Qin YM, Liu XH, Jia QL (2018) Synthesis of bamboo-like 3C–SiC nanowires with good luminescent property via nano–ZrO2 catalyzed chemical vapor deposition technique. Ceram Int 44:22890–22896. https://doi.org/10.1016/j.ceramint.2013.01.024
Liu ZL, Deng CJ, Yu C, Wang X, Ding J, Zhu HX (2018) Preparation of in situ grown silicon carbide whiskers onto graphite for application in Al2O3–C refractories. Ceram Int 44:13944–13950. https://doi.org/10.1016/j.ceramint.2018.04.243
Deng X, Li XC, Zhu BQ, Chen PG (2015) In-situ synthesis mechanism of plate-shaped β–SiAlON and its effect on Al2O3–C refractory properties. Ceram Int 41:14376–14382. https://doi.org/10.1016/j.ceramint.2018.04.243
Yin CF, Li XC, Chen PG, Zhu BQ (2019) Thermo-mechanical properties of Al2O3–C refractories with in situ synthesized non-oxide bonding phases. Ceram Int 45:7427–7436. https://doi.org/10.1016/j.ceramint.2019.01.032
Ma SH, Shi K, Xia Y (2020) Effect of modified tabular alumina aggregates on mechanical properties and microstructure of Al2O3–Al–C material. Ceram Int 46:9773–9779. https://doi.org/10.1016/j.ceramint.2019.12.249
GB/T 2997–2015 (2015) Test method for bulk density,apparent porosity and true porosity of dense shaped refractory products. https://d.wanfangdata.com.cn/standard/ChRTdGFuZGFyZE5ld1MyMDIxMDkwMhIOR0IvVCAyOTk3LTIwMTUaCGRzZDc0bWUx
GB/T 3001–2017 (2017) Refractory products-Determination of modulus of rupture at ambient temperature. https://d.wanfangdata.com.cn/standard/ChRTdGFuZGFyZE5ld1MyMDIxMDkwMhIOR0IvVCAzMDAxLTIwMTcaCHQ5cXBybGRn
GB/T 3002–2017 (2017) Refractory products-Determination of modules of rupture at elevated temperatures. https://d.wanfangdata.com.cn/standard/ChRTdGFuZGFyZE5ld1MyMDIxMDkwMhIOR0IvVCAzMDAyLTIwMTcaCHZ3eDNvNjFp
Chen ZY (2005) Chemical thermodynamics of refractories. Metallurgical Industry Press, Beijing
Yamaguchi A (1993) Practical thermodynamics and its application in high temperature ceramics. Wuhan Industrial University Press, Wuhan
Wu J, Bu NJ, Li HB, Zhen Q (2017) Effect of B4C on the properties and microstructure of Al2O3–SiC–C based trough castable refractories. Ceram Int 43:1402–1409. https://doi.org/10.1016/j.ceramint.2016.10.101
Zhang ZY, Yan W, Li N, Schafföner S, Zhou WY, Chen Z, Wei JW (2020) Mullite-corundum gas-permeable refractories reinforced by in-situ formed SiC whiskers. Ceram Int 46:25155–25163. https://doi.org/10.1016/j.ceramint.2020.06.302
An JC, Ge TZ, Xu EX, Zhao F, Liu XH (2019) Preparation and properties of mullite-SiC-O’-SiAlON composites for application in cement kiln. Ceram Int 46:15456–15463. https://doi.org/10.1016/j.ceramint.2020.03.090
Acknowledgements
This work had received financial support from the Scientific and Technological Research Project of Henan Provincial Department of Science and Technology (No. 212102210579).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors of this manuscript, including Xiaoyuan Han, Kai Shi, Shihang Ma, Yi Xia, Yang Liu, Jianzhao Shang, declare that no conflict of interests exists.
Additional information
Handling Editor: David Cann.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Han, X., Shi, K., Ma, S. et al. Effect of silicon powder-modified tabular alumina aggregates on the mechanical properties and microstructures of carbon-bonded alumina refractories. J Mater Sci 57, 914–924 (2022). https://doi.org/10.1007/s10853-021-06759-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-021-06759-4