
Abstract
Palladium nanoparticles (Pd NPs) supported on the surface of sodium dodecyl sulfonate (SDS)-intercalated layered double hydroxide (LDH) nanocomposites were synthesized by a one-step, facile ultrasonic method. The Pd/SDS–LDH nanocomposites were characterized by UV–visible spectroscopy, X-ray diffraction, transmission electron microscopy, scanning electron microscopy, X-ray photoelectron spectroscopy, N2-adsorption, Fourier transform infrared spectroscopy, and inductively coupled plasma optical emission spectrometry. The Pd NPs had an average size of 3.56 nm and were uniformly dispersed on the SDS–LDHs surface. The conversion of 4-bromotoluene catalyzed by Pd0.02/SDS–LDHs reached 98.16 % with 0.1 mmol % catalyst at room temperature without any phase transfer agents, toxic solvents, or inert atmosphere; this conversion was much higher than that of Pd0.02/SDS–LDHs prepared without ultrasound. This was attributed to the high dispersion and size uniformity. Notably, Pd0.05/SDS–LDHs had a much higher catalytic activity than that of commercial Pd/C catalyst with the same Pd content due to the strong interaction between the Pd species and the SDS–LDHs in the Pd/SDS–LDH nanocomposites. These catalysts could be easily separated by centrifugation, and could be recycled five times with little activity loss.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The palladium-catalyzed Suzuki–Miyaura coupling reaction [1, 2] is one of the most important organic transformations for the construction of carbon–carbon bonds, and it has been extensively used to synthesize natural products [3–5], pharmaceuticals [6], and advanced functional materials [7].
However, the traditional C–C coupling reactions have several disadvantages. First, toxic, sensitive organic solvents, phase transfer reagents, and complex organic ligands are used, and these compounds may cause serious environmental problems [8, 9]. Second, reactions are carried out in harsher reaction conditions, e.g., at higher temperatures and with inert gas, which consume more energy and generate more harmful gases [10, 11]. Furthermore, homogeneous palladium catalysts, which are the most widely used in Suzuki–Miyaura reactions, have an inevitable drawback that restricts their practical application, i.e., the difficulty in separating and recycling the expensive Pd species [12].
Heterogeneous catalysts are preferred over homogeneous catalysts due to their facile separation from the reaction mixture and the possibility of recycling. Supported Pd NPs play a pivotal role in a wide range of catalysis reactions, especially in Suzuki–Miyaura-type reactions. Nevertheless, there are also a number of obstacles, such as aggregation and leaching of Pd NPs on the surface of supports, which result in Pd having a low utilization efficiency. One solution for this problem is to uniformly disperse Pd NPs on the surface of supports, such as metal oxides [13, 14], carbon materials [15], porous materials [16], and polymers [17].
Recently, layered double hydroxides (LDHs), also known as hydrotalcites or hydrotalcite-like compounds, have attracted an increasing amount of attention as a new support for the most favorable layered crystals used in the preparation of nanocomposites; this attention is due to specific properties of LDHs, such as their ability to exchange anions, and to undergo reconstruction (also called the “structure memory effect”) [18, 19]. LDHs could be represented by the general formula [M(II)1−x M(III) × (OH)2]x+(A n−) x/n ·mH2O, where M (II) and M (III) represent any divalent and trivalent cations, respectively, and A n− is the interlayer anion [18, 19]. LDHs represent a type of natural or synthetic porous material that has a large surface area and many hydroxyl groups on the surface, which render these materials excellent options for dispersing and carrying Pd NPs. Anionic dodecyl sulfate (SDS) has been intercalated into the interlayers of LDHs mainly via anion exchange and has also been examined as a support for the improvement of Pd NPs dispersion and organic pollutant adsorption [18, 20].
Effectively controlling and stabilizing Pd NPs size and morphology is an alternative way to address the problem of low efficiency. Considerable attempts have been made to obtain uniform NP dispersions with effective control over particle size; these NPs are usually fabricated by physical and chemical reduction methods. Notably, the probability of NP aggregation increases as particles are reduced. To prevent undesired agglomeration, metal NP fabrication processes are often performed with the addition of a stabilizing reagent, such as functionalized polymers [21], surfactants [22], and tetraalkylammonium salts [23, 24]. However, one of the drawbacks of using stabilizers is that strong adsorption to the active sites of nanoparticles may diminish catalytic activity.
Sonochemical treatment is a new technology that has shown to be effective for preparing noble metals NPs. The decomposition of water molecules into hydrogen and hydroxyl radicals, which is induced under ultrasonic irradiation by high temperatures and high pressures generated in collapsing cavities, can be utilized to reduce high-quality nanoparticles [25–27]. Extensive studies have reported that Pd catalysts can be prepared by ultrasonic methods. For example, Tang et al. synthesized porous palladium nanostructures by sonicating a solution of K2PdCl4 and ascorbic acid for formic acid electro-oxidation [26]. Su et al. prepared Pd NPs supported on a g-C3N4 surface by ultrasonication [27]. However, in most cases, a relatively strong reductant (e.g., NaBH4, ascorbic acid), a long reaction time, or a specific, high-power ultrasonic system is needed. Furthermore, protective reagents or surfactants have been required in previous ultrasound synthesis processes [28, 29].
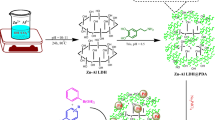
In this work, we present a facile and green synthesis method for fabricating Pd/SDS–LDH nanocomposites by ultrasonically treating Pd precursors in ethylene glycol. The Pd NPs dispersed uniformly on the surface of the SDS–LDHs without any additives or surfactants. The as-prepared catalyst exhibited superior catalytic performance in Suzuki–Miyaura coupling reactions, even in mild conditions, i.e., without phase transfer agents, toxic solvents, high temperatures or pressure or inert atmosphere. The catalyst can be reused up to six times without a significant loss of activity.
Experimental
Materials
All chemicals were of reagent grade and were purchased from Sinopharm Chemical Reagent Co., Ltd. All halides and phenylboronic acid (C6H7BO2) used for the Suzuki coupling reactions were purchased from Yurui Chemical Co., Ltd. (Shanghai). All the chemicals were used without further purification.
Synthesis of SDS–LDHs
Sodium dodecyl sulfate (SDS)-intercalated, layered double hydroxides (LDHs) [Mg3Al(OH)8]+(DS−)·2H2O were synthesized by a co-precipitation method. Typically, MgCl2·6H2O (12.198 g, 60 mmol), AlCl3·6H2O (14.484 g, 20 mmol), and SDS (5.7676 g, 20 mmol) were dissolved in deionized water (40 mL) to obtain solution (a) with Mg2+/Al3+/SDS at a molar ratio of 3:1:1, and an aqueous solution (b) containing NaOH (1.2 mol L−1) was prepared. Then, solution (b) was dripped into solution (a) to maintain the pH of the mixture at 9–10 under flowing N2. The obtained solution was transferred to a Teflon-lined stainless steel autoclave for a hydrothermal treatment at 100 °C for 16 h. After cooling, the resultant mixture was centrifuged and subsequently filtered. The filtrate was washed with deionized water three times and dried at 80 °C for 4 h, yielding SDS-intercalated MgAl-LDHs (denoted here as SDS–LDHs); the yield was = 7.22 g (67.4 %).
Synthesis of Pd/SDS–LDH nanocomposites
In a typical ultrasonic synthesis of Pd/SDS–LDH nanocomposites, 0.5 g of SDS–LDHs in 30 mL of ethylene glycol was mixed using an ultrasonic bath at a frequency of 25 kHz and an input power of 100 W for 30 min. The reaction temperature was maintained at 30 ± 1 °C by water surrounding the reactor. Next, 1.8 mL of the aqueous Na2PdCl4 (0.5 mol L−1) solution was rapidly added to the container and mixed under 25 kHz ultrasonic waves with an input power of 400 W for 30 min. The final black solid, denoted here as Pd0.02/SDS–LDHs, was centrifuged and washed with deionized water three times, subsequently dried at 70 °C in an oven overnight, and ground into a fine powder. For comparison, Pd0.005/SDS–LDH, Pd0.05/SDS–LDH and Pd0.10/SDS–LDH nanocomposites were also prepared under the same experimental conditions; the yield was 83.5, 84.3, 80.0, and 80.0 %, respectively. As a control, Pd/SDS–LDH nanocomposites were prepared by conventional magnetic stirring in the absence of ultrasound and are denoted here as “Pd/SDS–LDHs-T.” A schematic of the experimental setup is shown in Fig. 1.

Schematic of the ultrasonic generator
Characterization
The ultrasonic reduction of Na2PdCl4 solution was monitored by UV–vis spectrometer (Shimadzu spectrophotometer, model UV 2450). X-ray diffraction (XRD) patterns were measured on a Bruker D8 Advance X-ray diffractometer using Cu irradiation (λ = 0.15418 nm) with the scanning speed of 0.5°s−1 from 5° to 80° (2θ). Transmission electron microscopy (TEM) images were recorded with a JEM-2100FX Transmission electron microscope operating at 200 kV. Scanning electron microscopy (SEM) analysis was performed using a Philips FEI Sirion microscope. Brunauer–Emmett–Teller (BET) surface area was obtained by nitrogen sorption experiments conducted at 77 K using a Quantachrome Autosorb Gas Sorption analyzer. X-ray photoelectron spectroscopy (XPS) analysis was conducted on a Kratos AXIS ULTRA DLD spectrometer with a monochromatic Al Kα (1486.6 eV) radiation. The C 1 s peak at 284.6 eV was used for calibration. Surface chemistry of the synthesized particles was studied with FTIR which was carried out on Nicolet 6700 spectrophotometer (Thermo Nicolet, USA). The Pd loadings were determined by inductively coupled plasma optical emission spectrometry (ICP-OES, PerkinElmer, Optima 5300DV).
Suzuki–Miyaura coupling reaction
In a typical run, Pd/SDS–LDHs catalyst (13.30 mg, 2.5 μmol) was added to a stirred solution of aryl halide (392.5 mg, 2.5 mmol), K2CO3 (691.0 mg, 5.0 mmol), and phenylboronic acid (365.8 mg, 3.0 mmol) in the mixed solution of EtOH/H2O (30 mL). The reaction mixture was stirred at room temperature. After completion of the reaction, the catalyst was separated by centrifugation and then washed with ethanol three times. The mixture was extracted with ethyl acetate three times. The organic layers were combined and evaporated under reduced pressure to obtain crude product which was purified afterward by column chromatography. The purity of the products was identified by HPLC analysis, and yields were calculated on the basis of aryl halides.
Results and discussion
UV–visible spectra
UV–visible spectra with a variable wavelength from 250 to 800 nm were used to detect whether bivalent palladium was completely reduced to zerovalent palladium. Figure 2 shows the absorption spectra for palladium colloidal suspensions of different Pd loadings, which were ultrasonically treated for 30 min. The absorption of the Na2PdCl4 solution was used as a reference sample for comparison. The absorption bands presented in the reference sample spectrum are attributed to the characteristic absorption of PdCl4 2− species [28, 30]. The absence of absorption peaks above 300 nm in all of the samples indicates that the initial palladium (II) species was adequately reduced. These results showed that the ultrasonic method could quickly reduce palladium, which is an advantage.

UV–visible spectra of the Na2PdCl4 solution and Pd/SDS–LDHs nanocomposite suspensions with different Pd loads after 30 min of ultrasonic irradiation
XRD analysis
The XRD patterns of the LDHs, SDS–LDHs, and Pd/SDS–LDHs at 2θ = 5°–80° are shown in Fig. 3. The LDH XRD patterns (Fig. 3a) showed a typical, well-ordered, layered structure with a basal spacing (003) of 7.9 Å [31]. In the case of the SDS–LDHs samples (Fig. 3b), the (003) peak position shifted lower, to 2θ = 7.1°, with a larger basal spacing (003) of approximately 12.2 Å (Table S1); these results indicated that the SDS successfully intercalated into the LDHs interlayers [32]. With a brucite layer that is 4.8 Å thick (Al/Mg-hydroxide sheet), the interlayer space was calculated to be approximately 7.4 Å, which was lower than the length of the alkyl chains of C12H25SO3 − anions (approximately 20.8 Å), suggesting that the intercalated SDS was in a flat or tilted arrangement [33].

XRD patterns of a LDHs, b SDS–LDHs, c Pd0.005/SDS–LDHs, d Pd0.02/SDS–LDHs, e Pd0.05/SDS–LDHs, and f Pd0.10/SDS–LDHs
The overall form of the XRD pattern of the Pd/SDS–LDH nanocomposites was similar to that of the pure SDS–LDHs (Fig. 3b). All the Pd/SDS–LDH nanocomposites with different loads had nearly the same basal spacing value (Table S1), indicating that the Pd NPs were not between the layers but were instead on the LDH surface (likely the basal surface or edges). Compared with the pure SDS–LDHs, the peak intensity of the Pd/SDS–LDH nanocomposites with different loads was significantly broader in shape and lower in intensity due to the random dispersion of Pd NPs on the SDS–LDH surface, forming a hybrid composite. In Fig. 3c and d, as the Pd NPs were too small and the amount of Pd present was too low to yield sharp XRD peaks, only a broad modulation of the most intense (111) line of metallic Pd is observed at 2θ = 40.1° for the samples with a Pd load of 5 wt% (Fig. 3e). Up to a Pd load of 10 wt% (Fig. 3f), prominent Bragg reflections at 2θ values of 40.1°, 46.1°, and 67.9° were observed, corresponding to the (111), (200), and (220) Bragg reflections of the face-centered cubic (fcc) structure of Pd (JPDS 87-0643) [34]. These results suggested the successful fabrication of Pd/SDS–LDH nanocomposites.
SEM and TEM analyses
The morphology of the SDS–LDHs was investigated, and the results are shown in Fig. 4a, b. These samples consisted of platelet-like sheets. The random stacking of the platelet-like particles resulted in interparticle mesoporosity, which was consistent with the “mesoporous structure” indicated by the N2 adsorption–desorption curves (Fig. 6). The EDS analysis showed that the SDS–LDHs comprised Mg, Al, and S, and the Mg/Al atomic ratio of the sample was 17.2:5.4, approximately equal to the theoretical Mg/Al ratio (3:1). This finding demonstrated the formation of SDS-embedded, layered LDHs.

SEM image (a), TEM image (b) and EDS profile (c) of SDS–LDHs
The dispersions of smaller Pd NPs (3.42 nm) in the Pd0.005/SDS–LDHs were not very uniform, because the palladium loading was too low (Fig. 5a, S1). Figure 5b clearly demonstrates that in Pd0.02/SDS–LDH nanocomposite, small and spherical Pd NPs were evenly distributed on the surface without any aggregation, even without the use of any protective reagents or surfactants. Based on the particle size distribution histograms (Fig. 5b), the average particle size of the Pd0.02/SDS–LDHs was 3.56 nm, and these samples exhibited a very narrow size distribution. The high-resolution transmission electron microscopy (HRTEM) patterns further indicated the Pd crystal plane was 0.224 nm, which was attributed to the lattice spacing of the (111) plane of metallic Pd [35]. As shown in Fig. 5c, d, at a high loading (Pd0.50/SDS–LDHs and Pd0.10/SDS–LDHs), the Pd NPs tended to aggregate, which may be a detrimental factor that would decrease catalytic activity [36]. From the size distribution histograms, when the palladium loads increased to 10 %, the average particle size increased to 5.80 nm. The EDS analysis of Pd0.02/SDS–LDHs (Fig. 5e) also confirmed the existence of Pd NPs.

TEM images and corresponding size distributions of the Pd0.005/SDS–LDHs (a), Pd0.02/SDS–LDHs (b) (HRTEM images (inset)), Pd0.05/SDS–LDHs (c), and Pd0.10/SDS–LDHs (d), EDS profile of the Pd0.02/SDS–LDHs (e), Pd0.02/SDS–LDHs prepared in the absence of ultrasound (f)
To further study the effects of ultrasound, conventional magnetic stirring method was used instead of ultrasonic method to fabricate the Pd0.02/SDS–LDHs. Figure 5f shows that agglomerated Pd NPs, rather than dispersed Pd NPs, were observed on the surface of the SDS–LDHs. This further demonstrated that the ultrasonic treatment was essential for the formation of the uniform, well-dispersed Pd/SDS–LDH nanocomposites. Considering the above results, subsequent analyses were conducted based on the Pd0.02/SDS–LDHs, as an example. For further comparison, a commercial Pd/C catalyst was also investigated, and TEM images and size distribution histograms are shown in Fig. S2.
The exact amounts of Pd in the SDS–LDHs were detected via ICP analysis. The contents of Pd in the Pd0.005/SDS–LDHs, Pd0.02/SDS–LDHs, Pd0.05/SDS–LDHs, and Pd0.10/SDS–LDHs were determined to be 0.32, 1.17, 4.01, and 9.54 wt%, respectively. All the results revealed that the Pd/SDS–LDH nanocomposites had been successfully synthesized by the ultrasonic method and that the small Pd NPs were uniformly distributed on the SDS–LDHs without any agglomeration at an appropriate Pd load.
N2 adsorption–desorption
Nitrogen adsorption–desorption experiments were performed on SDS–LDHs and Pd0.02/SDS–LDHs (Fig. 6; Table 1). The characteristic isotherm corresponded to a type IV isotherm with a type H3 hysteresis loop, according to the IUPAC classification [28], exhibiting characteristics of interparticle mesoporosity created by the platelet-like particles. Such pores have been described as a “house of cards” structure [31, 37], which was consistent with the TEM observations (Fig. 4).

N2 adsorption–desorption isotherms of SDS–LDHs and Pd0.02/SDS–LDHs
Compared with the support SDS–LDHs, the catalyst Pd0.02/SDS–LDHs had a slightly lower surface area, as they were 101.991 and 91.197 m2g−1, respectively. In addition, the pore size of the Pd0.02/SDS–LDHs increased from 15.007 to 20.600 nm, as calculated from Barrett–Joyner–Halenda desorption isotherms (Table 1). These changes may have been due to ultrasonic effects, which could exfoliate the SDS–LDHs, resulting in a lower surface area and larger pore size during the synthesis of the catalyst. After supporting the Pd NPs, the pore volume of the Pd0.02/SDS–LDHs slightly decreased from 0.487 to 0.464 cm3g−1 because the Pd NPs became mostly dispersed in the “mesoporous structure.”
XPS analysis
Figure 7 shows a Pd 3d high-resolution XPS spectrum of the Pd0.02/SDS–LDHs. The Pd 3d3/2 peak at 341.55 eV and the Pd 3d5/2 peak at 336.25 eV were assigned to Pd (0), while the other doublet peaks at 343.35 and 337.95 eV were related attributed to Pd (II) [27, 28]. The Pd (II) present in the catalyst may come from an incomplete reduction of Pd (II) ions during the ultrasonic reaction and the formation of PdO by the oxidation of naked metal Pd atoms under ambient conditions. The doublet peaks in the Pd0.02/SDS–LDHs were slightly higher than the normal value of Pd 3d5/2 and Pd 3d3/2 [38]. This shift indicated that the chemical environment of the Pd had changed. This change could have been due to a portion of the electrons on the Pd surface being transferred to the positively charged LDH sheets, or Pd could have coordinated with other atoms and lost electrons, decreasing the electron density. These changes suggested that the Pd0.02/SDS–LDH nanocomposites are not mixtures of two unrelated compounds but rather are combined by strong metal-support interactions or chemical bonds, which are much more beneficial for catalyst dispersion, stability, and performance.

Pd 3d spectrum of the Pd0.02/SDS–LDHs
FTIR spectroscopy
An FTIR analysis was performed to study the changes in surface functional groups and to identify the functional groups potentially responsible for the reduction of the metal precursors and the stabilization of the supported Pd NPs in the ultrasonic synthesis process. In the range of 3800–2600 cm−1 (Fig. 8a), the broader band for the SDS–LDHs at 3474 cm−1 was ascribed to the stretching vibrations of OH in absorbed water. These vibrations shifted to 3491 cm−1 in the Pd0.02/SDS–LDHs, indicating that the surface properties of the SDS–LDHs had changed [39]. In the range of 400–2000 cm−1 (Fig. 8b), the SDS–LDHs showed the bending vibrations of OH groups forming hydrogen bonds (H–OH) at 1648 cm−1; this peak shifted to 1640 cm−1 in the Pd0.02/SDS–LDHs. The shifts of both the OH stretching (Fig. 8a) and H–O–H bending vibrations (Fig. 8b) influence the properties of the resulting materials [40].

FTIR spectra of SDS–LDHs and Pd0.02/SDS–LDHs in the range of a 3800–2600 cm−1 and b 2000–400 cm−1
Figure 8a displays two prominent vibrations at 2918 and 2849 cm−1, corresponding to the antisymmetric and symmetric –CH2– stretching modes, respectively. The symmetric and antisymmetric C–H stretching modes of the terminal –CH3 groups, existing in sodium dodecyl sulfate (SDS), at 2873 and 2955 cm−1 hardly disappeared in the Pd0.02/SDS–LDHs, and this may have resulted from the ultrasonic irradiation-induced transformation of the terminal –CH3 groups into secondary radicals. By the same token, the bending and stretching vibrations of alkane C–H groups at 1469, 1414, and 1379 cm−1 in the Pd0.02/SDS–LDHs became weaker than those of the SDS–LDHs.
The bands at 800 and 722 cm−1 were ascribed to the C–S–O and O–S–O bending vibrations of SDS (Fig. 8b). The C=O vibration located at 1743 cm−1, in the Pd0.02/SDS–LDHs spectrum, might be due to ultrasonic effects on the molecules in solution, such as sodium dodecyl sulfate (SDS) (Fig. 8b).
In particular, there was one broad band at 1175 cm−1 in the SDS–LDHs, which corresponded to the characteristic stretching vibration of S=O/S–O bonds of –OSO3 groups; this band further demonstrated the formation of SDS-intercalated LDHs. However, in the case of Pd0.02/SDS–LDHs, this band was split into two peaks, one at 1171 and at 1203 cm−1, indicating a decrease in the symmetry of the sulfate group [18, 41]. The decrease in symmetry revealed that three oxygens had different interactions with the environment, which further confirmed a reliable fixation of the Pd species. The bands at 800 and 722 cm−1 were ascribed to the bending vibrations of C–O–S and O–S–O bonds in the SDS.
In general, the bands in the range of 700–400 cm−1 were mainly ascribed to M–O lattice vibrations (M = Mg2+, Al3+), which were present in all the SDS–LDH and Pd0.02/SDS–LDH samples [41].
Mechanism of Pd/SDS–LDH synthesis
The ultrasound-induced reduction of Pd (II) to Pd (0) originated from acoustic cavitation, i.e., the formation, growth, and implosive collapse of bubbles in a liquid [42]. The collapsing cavities produced high-temperature and high-pressure conditions, leading to the decomposition of water molecules into hydrogen ·H and hydroxyl ·OH radicals, which could then react with HOCH2CH2OH to produce secondary radicals HOCH2C·HOH [36, 43]. The ultrasonic reduction of Pd (II) ions could occur according to the following steps (Eqs. (1–6)):
The overall procedure for synthesizing Pd/SDS–LDH nanocomposites is shown in Fig. 9. In summary, the proposed mechanism of the enhanced catalytic activity of Pd/SDS–LDH nanocomposites consists of the following factors: (1) the external hydroxyl groups of the SDS–LDHs promoted the production of more ·OH radicals in the presence of ultrasound, which supported reduction and dispersion of Pd (II); (2) the rich hydroxyl groups on the external surface of the SDS–LDHs could act to effectively disperse and fix the Pd species, strongly interacting between Pd species and support, both of which would affect the mobility of the species and the rate of nuclei growth, consequently influencing the catalytic activity; (3) the SDS-intercalated LDHs were selected as a support to enlarge the surface area and to improve the lipophilicity of the catalyst, which favors the attack of organic substrate molecules toward the catalyst; and (4) the interparticle mesoporosity created by the platelet-like sheets in the SDS–LDHs was conducive for the transmission of organic molecules, which is crucial for catalytic activity [44, 45].

Schematic representation of the Pd/SDS–LDH nanocomposites
Suzuki–Miyaura coupling reaction
Catalytic activity evaluation and kinetics study of the Pd/SDS–LDHs catalysts
The Suzuki–Miyaura reaction of 4-bromotoluene with phenylboronic acid was chosen to compare the catalytic performance of the synthesized Pd/SDS–LDHs (0.5, 2, 5, and 10 wt%), and the Pd0.02/SDS–LDHs prepared in the absence of ultrasound.
Figure 10 shows the yields at different intervals with various catalysts. No conversion of 4-bromotoluene was observed in the presence of only the support SDS–LDHs. Among all the catalysts with different Pd loads, the Pd0.02/SDS–LDHs exhibited the best and fastest catalytic activity; the conversion of 4-bromotoluene reached to 95.69 % at 30 min and, up to 98.16 % at 60 min with 0.1 mmol % catalyst at room temperature. These results were much higher than those of the Pd0.02/SDS–LDHs prepared in the absence of ultrasound. High Pd loads may not only lead to increased production cost, but also to lower catalytic activity resulting from increased particle size and aggregation, as supported by the TEM analysis. As shown in Fig. 5c, d, the Pd0.05/SDS–LDHs and Pd0.10/SDS–LDHs both exhibited aggregation to some extent. Thus, dispersion and size uniformity were the key factors in determining the activity of the catalysts. Further examination of the catalytic results revealed that under the current experimental conditions, the Pd0.05/SDS–LDHs had a much higher catalytic activity than did the commercial Pd/C catalyst that contained the same amount Pd. This result was ascribed to the relatively strong interaction between the Pd species and the SDS–LDHs in the Pd/SDS–LDH nanocomposites compared with the simple adsorption that occurs on the carbon surface of the Pd/C catalyst (as explained in 3.8).

Yields of biphenyl at different intervals with various catalysts
In order to elucidate the reaction kinetic process, a pseudo-second-order kinetic model was employed to fit the experimental data, as shown in Fig. 11, which can be expressed as follows:
where C A,0 and C B,0 (mol dm−3) are the initial concentrations of phenylboronic acid and 4-bromotoluene, and C A,t and C B,t (mol dm−3) are the concentrations of reactive phenylboronic acid and 4-bromotoluene, respectively, at the reaction time t. The reaction rate constants (k, mol dm3 min−1) of the various catalysts are shown in Fig. 11; these values further demonstrate that the Pd0.02/SDS–LDH nanocomposite was the most efficient catalyst because it exhibited the largest reaction rate constants (Table 2). Additionally, we calculated the intrinsic TOF, i.e., moles of 4-bromotoluene converted per mole of surface Pd per second, to further elucidate the distinct advantage of the Pd0.02/SDDS–LDHs. All the relevant values are shown in Table 2.

Kinetics curves of the Suzuki–Miyaura reaction with various catalysts

To optimize the reaction conditions, the effect of the solvents and base was examined, and the results are summarized in Tables S2 and S3. According to the optimization results and the principle of green chemistry, EtOH/H2O (v/v = 1:1) and 2.0 equiv of K2CO3 were chosen as the optimal conditions.
Under the optimized reaction conditions, to increase the applicability of the catalyst, the Suzuki–Miyaura reactions of different aryl halides with phenylboronic acid were studied. The results are shown in Table 3. Various p substituted aryl bromides, bearing either electron-donating or electron-withdrawing groups, such as –H, –NO2, –CN, –COCH3, –OCH3, and –CH3, underwent the cross-coupling smoothly and produced good yields of the corresponding products in mild conditions (Table 3, entries 1–6). Due to the steric hindrance effect, the o-substituted aryl bromides exhibited moderate yields (Table 3, entries 7–9).
To explore catalyst heterogeneity, the possible presence of dissolved palladium was investigated. After approximately 35 % of the coupling reaction had completed, the hot-filtered mother liquor was reacted with fresh substrates. No product was detected even after 1 h (Fig. 12). Moreover, an ICP analysis of the hot-filtered mother liquor and the catalyst after the reaction indicated that less than 0.1 % of the Pd species had leached out during the reaction (Table S4).

Suzuki coupling reaction in the presence of the hot-filtered mother liquor
The reusability of a catalyst is essential from an economic standpoint and for industrial applications. These catalysts could be recycled simply by centrifugation. In addition, the yield was almost unaltered over five cycles, but it decreased to 86.3 % (Fig. 13) in the sixth cycle.

The reusability of the Pd0.02/SDS–LDH catalyst for the Suzuki–Miyaura coupling reaction
To determine the reason of the decreased catalytic performance, the composition and structure of the fresh and recycled catalysts were examined. The XRD results showed that the primary layered structure was maintained in the catalyses recycled 1 and 6 times, while some metal palladium species were observed in the catalyst recycled 6 times (Fig. S3). As shown in Table S2, less than 0.1 % of leaching Pd was observed in the catalyst recycled once, and a Pd content of 0.94 wt% was found for the catalyst recycled 6 times, indicating that the Pd leached from the Pd0.02/SDS–LDHs during the reactions. As shown in Fig. 14, after 1 cycle, the Pd0.02/SDS–LDHs remained small and uniform particles, which was in contrast with the aggregated particles that were observed after 6 cycles. It can be concluded that the reduced catalytic activity was due to the Pd leaching and agglomerating.

TEM images of catalyst recycled 1 time (a) and 6 times (b)
Conclusion
In conclusion, an effective and reusable Pd/SDS–LDH nanocomposite was designed and successfully prepared by ultrasonically treating a Na2PdCl4 precursor solution in ethylene glycol. This ultrasonic approach provided fast reaction time and enabled catalyst synthesis under mild conditions without the use of any additives or surfactants. The Pd NPs were uniformly dispersed on the SDS–LDH surface, and the Pd/SDS–LDH nanocomposites exhibited excellent performance in Suzuki–Miyaura coupling reactions at room temperature without any phase transfer agents, toxic solvents, or inert atmosphere; thus, green catalysis was achieved. The high catalytic activity observed can be attributed to the intimate interaction between the Pd NPs and the support. The rich hydroxyl groups in the surface of the LDHs favored the formation of well-dispersed Pd NPs. This ultrasonic method provides a simple and environmentally friendly method for synthesizing Pd-supported catalysts and is a promising means of developing other advanced materials.
References
Miyaura N, Yamada K, Suzuki A (1979) Review: a new stereospecific cross-coupling by the palladium-catalyzed reaction of 1-alkenylboranes with 1-alkenyl or 1-alkynyl halides. Tetrahedron Lett 20:3437–3440
Miyaur N, Suzuki A (1995) Review: palladium-catalyzed cross-coupling reactions of organoboron compouds. Chem Rev 95:2457–2483
Kozlowski MC, Morgan BJ, Linton EC (2009) Review: total synthesis of chiral biaryl natural products by asymmetric biaryl coupling. Chem Soc Rev 38:3193–3207
Shen X, Jones GO, Watson DA, Bhayana B, Buchwald SL (2010) Review: enantioselective synthesis of axially chiral biaryls by the Pd-catalyzed Suzuki–Miyaura reaction: substrate scope and quantum mechanical investigations. J Am Chem Soc 132:11278–11287
Enthaler S (2011) Review: palladium-catalysed hydroxylation and alkoxylation. Chem Soc Rev 40:4912–4924
Tagata T, Nishid M (2003) Review: palladium charcoal-catalyzed Suzuki–Miyaura coupling to obtain arylpyridines and arylquinolines. J Org Chem 68:9412–9415
Ackermann L, Barfüsser S, Pospech J (2010) Review: palladium-catalyzed direct arylations, alkenylations, and benzylations through C–H bond cleavages with sulfamates or phosphates as electrophiles. Org Lett 12:724–726
Fihr A, Bouhrara M, Nekoueishahrak B, Basseta JM, Polshettiwar V (2011) Review: nanocatalysts for Suzuki cross-coupling reactions. Chem Soc Rev 40:5181–5203
Liu C, Ni Q, Bao F, Qiu JS (2011) Review: a simple and efficient protocol for a palladium-catalyzed ligand-free Suzuki reaction at room temperature in aqueous DMF. Green Chem 13:1260–1266
Fortman GC, Nolan SP (2011) Review: N-heterocyclic carbene (NHC) ligands and palladium in homogeneous cross-coupling catalysis: a perfect union. Chem Soc Rev 40:5151–5169
Metin Ö, Durap F, Aydemir M, Özkar S (2011) Review: palladium (0) nanoclusters stabilized by poly (4-styrenesulfonic acid-co-maleic scid) as an effective catalyst for Suzuki–Miyaura cross-coupling reactions in water. J Mol Catal A: Chem 337:39–44
Pagliar M, Pandarus V, Ciriminna R, Beland F, Demma Carà P (2012) Review: heterogeneous versus homogeneous palladium catalysts for cross-coupling reactions. Chem Cat Chem 4:432–445
Feng GF, Liu FJ, Lin C, Li W, Wang S, Qi C (2013) Review: crystalline mesoporous γ-Al2O3 supported palladium: novel and efficient catalyst for Suzuki–Miyaura reaction under controlled microwave heating. Catal Commun 37:27–31
Kumbhar A, Jadhav S, Kamble S, Rashinkar G, Salunkhe R (2013) Review: palladium supported hybrid cellulose-aluminum oxide composite for Suzuki–Miyaura cross coupling reaction. Tetrahedron Lett 54:1331–1337
Siamaki AR, Lin Y, Woodberry K, Connellc JW, Frank Gupton B (2013) Review: palladium nanoparticles supported on carbon nanotubes from solventless preparations: versatile catalysts for ligand-free Suzuki cross coupling reactions. J Mater Chem A 1:12909–12918
Okumura K, Tomiyama T, Okuda S, Yoshida H, Niwa M (2010) Review: origin of the excellent catalytic activity of Pd loaded on ultra-stable Y zeolites in Suzuki–Miyaura reactions. J Catal 273:156–166
Harish S, Mathiyarasu J, Phani KLN, Yegnaraman V (2009) Review: synthesis of conducting polymer supported Pd nanoparticles in aqueous medium and catalytic activity towards 4-nitrophenol reduction. Catal Lett 128:197–202
Zhang P, Qian G, Xu ZP, Shi H, Ruan X, Yang J, Frost RLP (2012) Review: effective adsorption of sodium dodecylsulfate (SDS) by hydrocalumite (CaAl-LDH-Cl) induced by self-dissolution and re-precipitation mechanism. J Colloid Interface Sci 367:264–271
Chuang YH, Liu CH, Tzou YM, Chang JS, Chiang PN, Wang MK (2010) Review: comparison and characterization of chemical surfactants and bio-surfactants intercalated with layered double hydroxides (LDHs) for removing naphthalene from contaminated aqueous solutions. Colloids Surf A 366:170–177
Gérardin C, Kostadinova D, Coq B, Tichit D (2008) Review: LDH nanocomposites with different guest entities as precursors of supported Ni catalysts. Chem Mater 20:2086–2094
Amali AJ, Rana RK (2009) Review: stabilisation of Pd (0) on Surface functionalised Fe3O4 nanoparticles: magnetically recoverable and stable recyclable catalyst for hydrogenation and Suzuki–Miyaura reactions. Green Chem 11:1781–1786
Uberman PM, Pérez LA, Lacconi GI, Martín SE (2012) Review: PVP-stabilized palladium nanoparticles electrochemically obtained as effective catalysts in aqueous medium Suzuki–Miyaura reaction. J Mol Catal A Chem 363:245–253
Upadhyay RK, Soin N, Saha S, Barman A, Roy SS (2015) Review: fast and facile preparation of CTAB based gels and their applications in Au and Ag nanoparticles synthesis. Mater Chem Phys 156:105–112
Fenger R, Fertitta E, Kirmse H, Thünemann AF, Rademann K (2012) Review: size dependent catalysis with CTAB-stabilized gold nanoparticles. Phys Chem Chem Phys 14:9343–9349
Darroudi M, Zak AK, Muhamad MR, Huang NM, Hakimi M (2012) Review: green synthesis of colloidal silver nanoparticles by sonochemical method. Mater Lett 66:117–120
Tang S, Vongehr S, Zheng Z, Ren H, Meng X (2012) Review: facile and rapid synthesis of spherical porous palladium nanostructures with high catalytic activity for formic acid electro-oxidation. Nanotechnol 23:255606–255616
Su X, Vinu A, Aldeyab SS, Zhong L (2015) Review: highly uniform Pd nanoparticles supported on g-C3N4 for efficiently catalytic Suzuki–Miyaura reactions. Catal Lett 145:1388–1395
Nemamcha A, Rehspringer JL, Khatmi D (2006) Review: synthesis of palladium nanoparticles by sonochemical reduction of palladium (II) nitrate in aqueous solution. J Phys Chem B 110:383–387
Nemamcha A, Moumeni H, Rehspringe JL (2009) Review: PVP protective mechanism of palladium nanoparticles obtained by sonochemical process. Phys Proced 2:713–717
Kan C, Cai W, Li C, Zhang L, Hofmeister H (2003) Review: ultrasonic synthesis and optical properties of Au/Pd bimetallic nanoparticles in ethylene hlycol. J Phys D Appl Phys 36:1609
Zhu J, Yuan P, He H, Frost R, Tao Q, Shen W, Bostrom T (2008) Review: in situ synthesis of surfactant/silane-modified hydrotalcites. J Colloid Interface Sci 319:498–504
Tao Q, Zhang Y, Zhang X, Yuan P, He H (2006) Review: synthesis and characterization of layered double hydroxides with a high aspect ratio. J Solid State Chem 179:708–715
He H, Frost RL, Deng F, Zhu J, Wen X, Yuan P (2004) Review: conformation of surfactant molecules in the interlayer of montmorillonite studied by 13C MAS NMR. Clays Clay Miner 52:350–356
Harraz FA, El-Hout SE, Killa HM, Ibrahim IA (2012) Review: palladium nanoparticles stabilized by polyethylene glycol: efficient, Recyclable catalyst for hydrogenation of styrene and nitrobenzene. J Catal 286:184–192
Song HM, Moosa BA, Khashab NM (2012) Review: water-dispersable hybrid Au–Pd nanoparticles as catalysts in ethanol oxidation, aqueous phase Suzuki–Miyaura and heck reactions. J Mater Chem 22:15953–15959
Hu B, Cai F, Chen T, Fan M, Song C, Yan X, Shi W (2015) Review: hydrothermal synthesis g-C3N4/nano-InVO4 nanocomposites and enhanced photocatalytic activity for hydrogen production under visible light irradiation. Appl Mater Int 7:18247–18256
Gursky Jennifer A, Blough Sandra D, Luna Cesar, Gomez Clarissa, Luevano Amber N, Gardner Elizabeth A (2006) Review: particle-particle interactions between layered double hydroxide nanoparticles. J Am Chem 128:8376–8377
Sun J, Fu Y, He G, Sun X, Wang X (2015) Review: green Suzuki–Miyaura coupling reaction catalyzed by palladium nanoparticles supported on graphitic carbon nitride. Appl Catal B 165:661–667
Tao Q, He H, Frost RL, Yuan P, Zhu J (2009) Review: nanomaterials based upon silylated layered double hydroxides. Appl Surf Sci 255:4334–4340
Hongping H, Ray FL, Jianxi Z (2004) Review: infrared Study of HDTMA+ intercalated montmorillonite. Spectrochim Acta Part A 60:2853–2859
Zhang P, Wang T, Qian G, Wu D, Frost RL (2015) Review: effective intercalation of sodium dodecylsulfate (SDS) into hydrocalumite: mechanism siscussion via near-infrared and mid-infrared investigations. Spectrochim Acta Part A 149:166–172
Okitsu K, Bandow H, Maeda Y, Nagata Y (1996) Review: sonochemical preparation of ultrafine palladium particles. Chem Mater 8:315–317
Okitsu K, Yue A, Tanabe S, Matsumoto HK (2000) Review: sonochemical preparation and catalytic behavior of highly dispersed palladium nanoparticles on alumina. Chem Mater 12:3006–3011
Rae J, Ashokkumar M, Eulaerts O, von Sonntag C, Reisse J, Grieser F (2005) Review: estimation of ultrasound induced cavitation bubble temperatures in aqueous solutions. Ultrason Sonochem 12:325–329
Lin Q, Li L, Liang S, Liu M, Bi J, Wu L (2015) Review: efficient synthesis of monolayer carbon nitride 2D nanosheet with tunable concentration and enhanced visible-light photocatalytic activities. Appl Catal B 163:135–142
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21276067), Natural Science Foundation of China- Russian Foundation for Basic Research (NSFC-RFBR, Grant No. 214111301884), Program of International S&T cooperation (Grant No. 2013DFR40570), and Science Foundation of Heilongjiang Academy of Sciences.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Li, J., Bai, X. Ultrasonic synthesis of supported palladium nanoparticles for room-temperature Suzuki–Miyaura coupling. J Mater Sci 51, 9108–9122 (2016). https://doi.org/10.1007/s10853-016-0164-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-016-0164-5