
Abstract
The effect of heating rate on the phase transformation kinetics of a Ti–10V–2Fe–3Al metastable β titanium alloy quenched from the β field is investigated by fast in situ high energy synchrotron X-ray diffraction and differential scanning calorimetry. The initial microstructure is formed by α″ martensite and fine ωath particles distributed in the retained β-phase matrix. The phase transformation sequence varies with the heating rate as revealed by analysis of the continuous evolution of crystallographic relationships between phases. At low temperatures an athermal reversion of α″ martensite into β takes place. This reversion occurs to a larger extent with increasing heating rate. On the other hand, diffusion–driven precipitation and growth of the ω phase is observed for lower heating rates accompanying the reverse martensitic transformation. Furthermore, the results show that the stable α phase can form through three different paths: (a) from the ω phase, (b) from α″ martensite, and (c) from the β phase.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Metastable β titanium alloys are mostly used in a wide range of components for the aerospace industry and as alternative structural materials for the automotive sector due to their high specific strength and excellent fatigue resistance [1, 2]. Moreover, biomedical applications are also being considered due to biocompatibility of β-Ti alloys and partial or total superelasticity [3]. The mechanical properties of these alloys for structural applications are basically defined by a homogeneous distribution of α/β interfaces due to the presence of fine α precipitates embedded in a β matrix [4]. On the other hand, the superelastic behavior is associated to a reverse martensitic transformation that leads to shape memory effect [5]. The formation of the α phase, its morphology, volume fraction, size, distribution as well as the progression of eventual shape memory effect are influenced by the phase transformation kinetics during heat treatments [6]. Therefore, a correct understanding of these processes is required for the optimization of the heat treatment and chemical composition of the alloys. In most of the prior works the study of phase transformations in metastable β titanium alloys was discussed on the basis of isothermal aging and ex situ experiments [7–11]. Although important insights have been gained using these methods, the role played by kinetic variables (e.g., heating rate) on the phase transformation sequence has received less attention [12–14]. Nowadays, continuous and univocal studies of phase transformation kinetics during heat treatment can be carried out in situ by high energy synchrotron X-ray diffraction (HEXRD) [15–17].
The Ti–10V–2Fe–3Al metastable β titanium alloy is extensively used in aircraft components such as landing gear structures or helicopter rotors [2, 18]. Depending on the thermomechanical process, this alloy can develop bi-modal, lamellar, or fine α plate microstructures that offer better combinations of tensile strength, fatigue resistance, and fracture toughness than α + β titanium alloys [18, 19].
In the present study, the phase transformations kinetics of a Ti–10V–2Fe–3Al alloy in β-quenched state are determined in situ as a function of the heating rate by means of HEXRD and complemented by differential scanning calorimetry (DSC). The evolution of stable and metastable phases is analyzed in terms of volume fraction and the crystallographic relationships between their lattice structures during linear heating from room temperature (RT) to the β-field.
Experimental
Material
The composition of the investigated metastable β titanium alloy Ti–10V–2Fe–3Al is given in Table 1. The β-transus temperature of the as-received primary ingot is 800 °C [20]. Samples of 20 × 20 × 30 mm3 were cut from the as-received ingot and subjected to an isothermal heat treatment at 900 °C (β-field) for 20 min in a dynamic argon atmosphere and quenched in RT water. This water-quenched condition corresponds to the initial condition for the further investigations.
Microstructural characterization
Specimens were prepared for light optical microscopy (LOM) by electropolishing for 35 s at RT in a LectroPol-5 machine using a solution of 8 % HClO4 + 12 % 2-butoxyethanol + 80 % ethanol and a voltage of 40 V. This procedure was effective in removing the presence of stress-induced α″ martensite formed during mechanical polishing [6]. Subsequently, the samples were immersed for 20 s in a Kroll’s reagent solution (2 % HF + 4 % HNO3 + 94 % H2O) to reveal the microstructural constituents. LOM was carried out using a Zeiss Axioplan optical microscope. The grain size was determined using the ASTM standard E 1382-91 as implemented in the software Axio Vision 4.8.2.
Thin-foil specimens were prepared for transmission electron microscopy (TEM) by dimpling followed by ion milling using a Gatan precision ion polishing system (PIPS) device employed at 4 kV. The samples were examined with a Tecnai F20 microscope operated at 200 kV.
Study of phase transformation kinetics
DSC and in situ HEXRD were used to determine the phase transformation kinetics of the Ti–10V–2Fe–3Al alloy. The evolution of phases was studied during continuous heating at constant heating rates of 5, 20, and 50 °C min−1 from RT up to 900 °C.
Differential scanning calorimetry
DSC was carried out in a Netzsch DSC 404C Pegasus calorimeter using specimens of ~5 mm diameter and ~1.6 mm thickness (~145 mg) cut using a SiC disk. A pure Al2O3 sample was used as a reference. The investigated samples and the reference were inserted into Al2O3 pans. The DSC experiments were performed in argon 5.0 atmosphere with a flow rate of ~80 ml min−1 to avoid oxidation. The DSC curves show a continuous sequence of reaction peaks that make it difficult to determine a reference baseline. Therefore, the start and finish temperatures of individual reactions were determined by the inflection points between them as described in [21, 22].
In situ high energy synchrotron X-ray diffraction
In situ HEXRD was carried out at two different synchrotron radiation sources: ID15B beamline of the European Synchrotron Radiation Facility (ESRF, Grenoble) [23] and P07-HEMS beamline at PETRA III, Deutsches Elektronen-Synchrotron (DESY, Hamburg) [24]. The experimental parameters for each setup are summarized in Table 2. Samples of 4 × 4 × 20 mm3 (ID15B) and 4 × 4 × 10 mm3 (P07) were cut using a SiC disk. The ID15B samples were heated up in a dynamic argon 4.6 atmosphere by means of a radiant furnace [16]. A modified dilatometer Bähr 805A/D with an induction coil furnace [25] was used at the P07-HEMS beamline to heat up the samples in a helium 5.0 atm. In both cases, the samples were kept fixed during acquisition and the temperature was controlled by a spot-welded thermocouple located next to the position of the incoming beam. Image sequences of complete Debye–Scherrer rings from the bulk of the samples were recorded in transmission mode (thickness = 4 mm) using image-plate detectors (see Table 2).
Analysis of diffraction data
The instrumental parameters of each setup were obtained using a LaB6 powder standard. Qualitative analysis of the evolution of the diffraction patterns during heating was carried out using two procedures:
-
(1)
Azimuthal integration of the intensity along the Debye–Scherrer rings at each temperature using the software Fit2d [26].
-
(2)
Unrolling the Debye–Scherrer rings and converting them into Cartesian coordinates (azimuthal angle, 2θ) and projection of the maximum intensity on the 2θ axis using the software ImageJ [27].
Quantitative phase analysis of the diffraction patterns, such as the evolution of volume fraction of phases and the cell parameters, was determined by the Rietveld method as implemented in the software Maud [28].
Results
Initial microstructure
Figure 1a shows a light optical micrograph of the initial microstructure of the Ti–10V–2Fe–3Al alloy. Large α″ martensite plates can be observed distributed at the grain boundaries of the retained β matrix. This is clearly shown in the inset of Fig. 1a where α″ martensite plates nucleated at the β grain boundaries can be observed. Moreover, the β grains present equiaxed morphology with a mean diameter of ~400 µm. Analogous microstructures have been reported for Ti–10V–2Fe–3Al quenched from the β-field [6, 7].

a LOM image of the initial microstructure formed by a matrix of metastable β grains surrounded by α″ martensite plates. b TEM bright field image of the initial microstructure showing fine laths and larger plates of α″ martensite. The inset in b presents the electron diffraction pattern of the \( \left[ {\bar{1}\bar{1}3} \right] \) β zone axis in the circled region and reveals the presence of ωath
TEM of the initial microstructure reveals that α″ martensite can also be found distributed in the β matrix as lath colonies and as plates (Fig. 1b). Figure 1b refers to a region located in the interior of β grains. In addition, irregularly shaped and very fine (1–10 nm) particles corresponding to the athermal ω phase (ωath) are also found within the β matrix and in between the α″ martensite. Particles of ωath with a mean size of ~3 nm have been reported for the current alloy after quenching from the β-field [7]. The inset in Fig. 1b shows the ωath and α″ phases identified by electron diffraction in the encircled region. (\( 1\bar{1}0 \)) spots of α″ appear next to those from (\( 1\bar{1}0 \)) of β [29]. Furthermore, characteristic features of ωath such as sharp spots and diffuse intensity distribution (diffuse ω [30], diffuse streaking [31]) can be observed. Similar electron diffraction patterns denoting the presence of ωath were reported in previous works [30–33]. The nature of diffuse scattering in ωath has been interpreted in terms of incoherent precipitates [34]. Moreover, it has been suggested that the ωath precipitates are formed to relax the strain caused by the volume expansion from the β phase to the α″ martensite during quenching [35, 36]. Therefore, it can be expected that the ωath precipitates are located at the α″–β interface, but this cannot be revealed with the spatial resolution obtained by TEM in this work.
Influence of heating rate on phase transformations during continuous heating
Differential scanning calorimetry
Figure 2 presents the DSC results obtained during continuous heating of the initial microstructure. The DSC curves show that the heat flow during the thermal treatment increases with the heating rate. The three curves present similar peak (transformation) sequences with a shift to higher temperatures with increasing heating rate. The endothermic transformations are indicated by numbers enclosed in circles, while the exothermic ones are shown by numbers in squares. Corresponding peaks at each heating rate are connected by grey lines. The 20 and 50 °C min−1 conditions present the same number of peaks (7). A shift of 15–50 °C is observed for the reactions taking place at 50 °C min−1 with respect to 20 °C min−1 heating rate. On the other hand, the 5 °C min−1 curve shows two exothermic transformations in the range between 380 and 435 °C (peak 4), and the transformation sequence is shifted 15–40 °C to lower temperatures with respect to the 20 °C min−1 condition. The endothermic peak 7 shows an asymmetric shape that may be due to an overlapping of two or more endothermic transformations. Except for this peak and the two peaks 4 at 5 °C min−1, each exothermic peak is followed by an endothermic transformation and vice versa, indicating formation and transformation of phases, respectively [13, 14, 37].

DSC heat flow curves obtained during continuous heating of the initial microstructure at 5, 20, and 50 °C min−1 up to 900 °C. The equivalent peaks between conditions are connected by grey lines. The peaks are numbered in ascending order during continuous heating. Peaks related to endothermic and exothermic reactions are indicated by circles and squares, respectively (Color figure online)
In situ high energy synchrotron X-ray diffraction
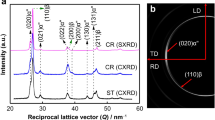
Portions of characteristic Debye–Scherrer rings obtained by HEXRD during heating at 5 and 50 °C min−1 can be observed in Fig. 3. They correspond to diffraction patterns at four different temperatures in order to illustrate the rings’ evolution. The images shown in Fig. 3a, b were obtained during experiments carried out at the beamlines P07-PetraIII and ID15B-ESRF, respectively. The diffuse ring observed near the center of Fig. 3b corresponds to the scattering of a quartz glass capsule used to keep the sample in inert gas atmosphere. The relatively large grain size of the initial microstructure is reflected by the spotty structure of the diffraction patterns at 25 °C. The diffraction rings of the 5 °C min−1 experiment appear more continuous than those of the 50 °C min−1 experiment due to the larger gauge volume investigated using the setup at P07 (see Table 2). At 370 °C, some Debye–Scherrer rings observed at 25 °C for 50 °C min−1 disappear while new reflections appear. On the other hand, all the rings observed at 25 °C for the 5 °C min−1 condition remain and some coarse spots as well as new weak reflections appear. The next two temperatures (450 and 600 °C) show basically the same diffraction rings with similar scattering angles. Furthermore, the diffraction pattern at 450 °C for the 50 °C min−1 condition shows streaks that link coarse grains to one or more low intensity spots with a straight line forming a cross-like shape (two of these regions are highlighted by circles in Fig. 3). These streaks indicate orientation relationships in the transforming region of the grain [38].

Quarters of representative Debye–Scherrer rings obtained at 25, 370, 450, and 600 °C during continuous heating: a 5 °C min−1 (P07-PetraIII) and b 50 °C min−1 (ID15B-ESRF)
The evolution with temperature of the integrated diffraction patterns is shown in Fig. 4a, b for a small 2θ range that represents qualitatively the complete HEXRD experiments for the 5 and 50 °C min−1 heating rates, respectively. It must be pointed out that the ratio between peak intensities is not representative of the volume fraction of the respective phases due to the small 2θ range depicted. The results for the 20 °C min−1 heating rate are between those for 5 and 50 °C min−1 conditions and have been excluded to improve legibility. The β and α″ phases are revealed by the diffraction patterns at 25 °C in all studied samples. No evidence of the ωath phase identified by electron diffraction in the transmission electron microscope could be obtained by HEXRD in the initial condition of the alloy. This may be due to an overlapping of the highest intensity ωath reflections with the more extensive β-peaks, and the weak and broad ωath reflections resulting from the diffuse scattering of this phase associated to lattice distortions [39]. The difficulty in identifying ωath in metastable β titanium alloys by X-ray diffraction has been reported by several authors [16, 35, 40].

Evolution of the diffraction patterns (limited to a small 2θ range) as a function of temperature during continuous heating up to 600 °C: a 5 °C min−1 (P07-PetraIII) and b 50 °C min−1 (ID15B-ESRF)
A sudden intensity decrease of the α″ reflections, followed by an increase in the intensity of the β-reflection can be observed between 200 and 260 °C. The α″ intensity drop is more pronounced for the 50 °C min−1 heating rate, where the α″ reflections practically vanish. This is a diffusionless reverse α″ → β transformation which is responsible for the one-way shape memory effect that is observed in the Ti–10V–2Fe–3Al alloy [6]. This can be seen more clearly in Fig. 5a, b where the Debye–Scherrer rings are presented in Cartesian coordinates for the 50 °C min−1 heating rate at 25 and 265 °C, respectively. The {hkl} indices of the reflections of the various phases are indicated. At 265 °C (Fig. 5b) the α″ reflections have vanished, while a pronounced and localized intensity increase of some β spots takes place. On the other hand, Fig. 6 shows the changes that take place in an equivalent temperature range for the 5 °C min−1 condition. Here, only a partial extinction of α″ is observed at 250 °C (Fig. 6b) as evidenced by an intensity reduction of the reflections of this phase. Also, the β reflections remain less coarse than for the 50 °C min−1 heating rate (Fig. 5b). This indicates that the reverse α″ → β transformation is more pronounced with increasing heating rate [6] and results in a full transformation of α″ into β at 50 °C min−1.

Complete Debye–Scherrer rings converted into Cartesian coordinates for the 50 °C min−1 heating experiment: a RT and b 265 °C, after reverse martensitic transformation

Complete Debye–Scherrer rings converted into Cartesian coordinates for the 5 °C min−1 heating experiment: a RT and b 250 °C
Another characteristic of the diffraction patterns shown in Figs. 5b and 6b is the emergence of low intensity spots that correspond to the ω phase. This is a thermally activated phase known as isothermal ω (referred to as ω in this work) (see [41]). Families of spots associated to {110}, {101}, {201}, {300}, {211}, and {102} reflections of ω lay next to the 2θ positions where the {110}, {200}, and {211} reflections of β are located. This overlapping causes the apparent fluctuations in the intensity of β observed in Fig. 4 at ~250 °C.
Figure 7a, b shows color-coded 2D plots corresponding to the evolution of several {hkl} reflections during continuous heating at 5 °C min−1 up to 600 °C for characteristic 2θ ranges. The reflections of the α″ phase are attenuated at ~250 °C owing to the partial reverse α″ → β transformation. In addition, the interplanar distance of the {020}α″, {021}α″, and {130}α″ decreases reaching a minimum at ~275 °C and then increases up to ~450 °C until approximately the same values as for RT are reached. On the contrary, the {110}α″, {111}α″, and {200}α″ reflections follow the opposite behavior. Additionally, at ~250 °C the intensity of the {201} reflection of ω increases. The {110}, {101}, {111}, and {002} of ω become distinguishable at ~325 °C. All ω reflections vanish at ~400 °C. At this temperature the α″ reflections start to shift towards the position of α while their intensity decreases. New reflections indicated by arrows slightly above ~400 °C appear next to {200}α″ and {130}α″. The α″ reflections disappear between 425 and 450 °C. Reflections corresponding to the stable α phase can be appreciated at T > ~525 °C.

Color-coded 2D plots corresponding to the evolution of several {hkl} reflections during continuous heating at 5 °C min−1 up to 600 °C for characteristic 2θ ranges: a 2.6–3.4° and b 3.8–5.1° (Color figure online)
The sudden vanishing of α″ reflections at ~200 °C due to the full reverse martensitic transformation into β can be clearly observed in Fig. 8 for 50 °C min−1 heating rate. Similar to 5 °C min−1, the reflections associated to ω become visible at ~350 °C and disappear slightly below ~450 °C. Also, new reflections (indicated by arrows) that do not correspond to α″ martensite, β, ω or α but are similar to those expected for α″ martensite start to form at this temperature. These reflections move towards the position of the α phase up to ~550 °C, where clear stable α phase reflections can be observed. A relationship between ω and the new reflections can be observed in Fig. 8b at 425 °C, where the {002}ω reflection shifts to a position close to that of {130}α″. Figure 9 shows a detail of the same {hkl} reflections as in Figs. 7b and 8b over a small azimuth range at three characteristic temperatures for the 5, 20, and 50 °C min−1 heating rates. At 25 °C, the reflections corresponding to α″ and β are present. At 400, 415, and 450 °C (ascending with the heating rate) the ω reflections start to disappear for each heating rate, showing that the reflection {002}ω shifts towards the position of {130}α″. This corresponds to the period at which the reflections indicated by arrows close to α″ become visible in Figs. 7b and 8b. Furthermore, streaking can be observed between the overlapping {201}ω and {200}β reflections, and the reflections {200}α″ and {022}α″. For the 20 and 50 °C min−1 conditions, extra spots between {200}α″ and {130}α″ (indicated by arrows) are visible. At 450, 455, and 490 °C (bottom figures) the streaking effect still remains for each heating rate and the {112}α″ with {022}α″ and {200}α″ with {130}α″ reflections tend to join. These evolutions can be visualized more clearly in the following video sequences: Online Resources 1, 2, and 3, respectively.

Color-coded 2D plots corresponding to the evolution of several {hkl} reflections during continuous heating at 50 °C min−1 up to 600 °C for characteristic 2θ ranges: a 3.0–4.0° and b 4.6–5.9° (Color figure online)

Color-coded 2D plots of selected portions of complete Debye–Scherrer rings converted into Cartesian coordinates. The evolution of the Bragg reflections is shown for three different temperatures: a 5 °C min−1, b 20 °C min−1, and c 50 °C min−1 (Color figure online)
Discussion
The phase transformations observed by in situ HEXRD were described qualitatively in the last section. Hereafter, these processes are studied by quantitative phase analysis using the Rietveld method as implemented in the software Maud [28]. This requires the knowledge of the crystallographic structure of the phases involved during the transformations, namely α, α″, ω, and β, which are summarized in Table 3. The structures of the metastable α″ and ω phases are described by the respective y and z ω parameters, which are directly related to the transformation mechanisms of these phases. The α″ phase is believed to be the result of an incomplete shear of the {110}β planes as they tend to transform into the basal planes of hexagonal α′ martensite during quenching or deformation of metastable β titanium alloys [42]. Due to its lower symmetry, the α″ phase is considered to be a disordered phase in which shuffle displacements from the transformation can be reflected in the atomic y-coordinate [30] (Table 3). An α hcp structure is obtained for \( y = 1/6,\;b/a = \sqrt 3 \), while for a β bcc structure \( y = 1/4,\;b/a = \sqrt 2 \) [43]. Possible α″ martensite structures lie between these values. In the current work, y ~ 0.186 was determined by Rietveld refinement for the initial microstructure of Ti–10V–2Fe–3Al. Prior studies showed that for Ti–Nb alloys the y-coordinate increases with the solute content [43–45]. This value lies between those found for the binary metastable β Ti–14Nb (y = 0.1810) and Ti–20Nb (y = 0.1877) alloys [44]. Higher values for the y-coordinate (~0.222) were reported for higher contents of Nb (~24 wt%) [43].
The formation of the ω phase consists of a collapse of two-thirds of the {111}β layers into double layers, whereas the other one-third remains as a single layer [30]. During the transformation, z ω varies continuously between 0 ≤ z ω ≤ 1/6. In this way, z ω = 0 corresponds to the bcc β phase, while z ω = 1/6 is the ideal ω phase with hexagonal symmetry. Structures with z ω between 0 and 1/6 are considered trigonal [41]. The relationship between the cell parameters of the β and the hexagonal ω phase can be expressed as \( a_{\omega } = \sqrt 2 a_{\beta } \) and \( c_{\omega } = \sqrt 3 /2a_{\beta } \) [46]. In the current work no intermediate states of the ω phase are considered in the Rietveld analysis since this assumption resulted in the best refinement of the diffraction patterns. However, it is important to remember that certain volume fraction of ωath was observed by diffraction analysis of a selected area in TEM (Fig. 1b).
Kinetics of the athermal α″ → β reversion
The phase transformations observed in the temperature range between 170 and 315 °C (see Figs. 5, 6, 7, 8) involve the α″, ω, and β phases. Figure 10 shows the evolution of the phase volume fractions as a function of temperature for the three different heating rates. At the beginning of the heating the 5 and 20 °C min−1 conditions present an α″ content of ~70–80 vol% while the 50 °C min−1 condition shows an initial content of ~60–70 vol%. As it will be pointed out later these differences may be due to slight differences in the distribution of quenching strains in the samples. The apparent difference in volume fraction of martensite with respect to the light optical micrograph in Fig. 1a can be understood taking into account that the martensite is also present embedded in the β-matrix as it is shown in Fig. 1b. At ~200 °C all heating rates present a fast α″ → β reversion whose magnitude increases with heating rate. Thus, the volume fraction of α″ martensite after reversion in the 5 and 20 °C min−1 conditions reaches a minimum of ~40 and ~15 vol%, respectively. On the other hand, the 50 °C min−1 condition shows a full transformation of α″ into β. Moreover, Fig. 11 presents the evolution of the cell parameters as a function of temperature for the three different heating rates. During heating from RT to the end of the α″ → β reversion (dashed grey lines in Fig. 11), b α″ and a α″ decrease and increase, respectively, while c α″ remains practically constant. These changes are more accentuated for the 20 °C min−1 condition between 250 and 300 °C (Fig. 11b). Furthermore, the temperature at which the minimum α″ volume fraction is reached for the 5 and 20 °C min−1 conditions at the end of the α″ → β reversion coincides with that of the minimum and maximum reached for the b α″ and a α″ cell parameters, respectively.

Evolution of the phase volume fractions as a function of the temperature during continuous heating: a 5 °C min−1, b 20 °C min−1, and c 50 °C min−1

Evolution of the cell parameters and of the crystallographic relationships between phases as a function of temperature during continuous heating: a 5 °C min−1, b 20 °C min−1, and c 50 °C min−1
The athermal α″ → β reversion is a diffusionless transformation caused by thermal mismatch stresses generated during heating from RT at the interface between α″ and β. This indicates that the athermal α″ → β transformation is controlled by lattice strains between these phases [6]. In the initial microstructure the three principal strains between α″ and β generated by the β → α″ transformation during quenching can be calculated as presented in [43]:
Slight variations are obtained at RT considering all initial states: ϵ 1 = −(7.1–7.5) %, ϵ 2 = +(7.0–7.1) %, ϵ 3 = +(0.6–0.8) %. These values are close to those presented for this alloy in similar conditions [6] and for a quenched β-metastable Ti–25Nb wt% alloy [47]. On the other hand, at the end of the athermal α″ → β reversion (dashed grey lines in Fig. 11) ϵ 1 ~ −6.3 %, ϵ 2 ~ 6 %, ϵ 3 ~ 0.8 % and ϵ 1 ~ −4.7 %, ϵ 2 ~ 5.2 %, ϵ 3 ~ 1.4 % for the 5 and 20 °C min−1 conditions, respectively, reveal a relaxation of ϵ 1 and ϵ 2 (a slight increase of ϵ 3 is also observed for 20 °C min−1). The evolution of strain magnitudes as the heating rate increases presents similar values to those observed in Ti–Nb alloys by increasing the solute content from ~25 to 32 wt% [47].
A continuous increase of the y-coordinate was obtained during heating until a maximum value is reached at the end of the athermal α″ → β reversion. These maximums are 0.194 and 0.198 for the 5 and 20 °C min−1 conditions, respectively, and, therefore, tend to be closer to the ones required to generate a bcc structure (y = 1/4).
Table 4 summarizes the temperature spans of the phase transformations shown in Fig. 10 for each heating rate, and the corresponding temperature ranges obtained for the DSC peaks (Fig. 2). It must be pointed out that the direction of the reaction (exo- or endothermic) indicated in Table 4 only refers to the dominating process and does not exclude the possibility that an overlapping of several reactions takes place simultaneously. The α″ → β reversion starts in the range of 170–200 °C and finishes between 260 and 315 °C. These values are close to the ones reported in [6] for the same alloy. The difference in the starting temperature and the temperature ranges during which the phase transformation takes place may be due to the different initial strain conditions ϵ 1, ϵ 2, and ϵ 3 generated during water quenching of the samples.
Formation of ω
The formation of the ω phase starts simultaneously with the athermal α″ → β reversion for all heating rates (Fig. 10). This leads to an overlapping of an endothermal (α″ → β) and an exothermal (ω formation) phase transformation in the DSC signal that results in the differences between the finish temperatures of the α″ → β transformation obtained by DSC and HEXRD (peak 1 in Table 4). For the β → ω transformation, both methods present similar temperature spans which increase with heating rate. Figure 10 shows that ω reaches maximum volume fractions of 22, 5, and 80 vol% for the 5, 20, and 50 °C min−1 heating rates, respectively. The considerably large ω volume fraction obtained for 50 °C min−1 is comparable with the ones reported (80–90 vol%) for other metastable β titanium alloys with similar content of β stabilizers (Ti–23Nb, Ti–10V, Ti–11Mo, Ti–8Mn, and Ti–6.8Mo–4.5Fe–1.5Al wt%) [36, 46, 48, 49].
The lower maximum ω volume fraction observed comparing 5 and 20 °C min−1, and the subsequent increase comparing 20 and 50 °C min−1, can be understood taking into account that two different mechanisms, depending on the heating rate, are responsible for the precipitation of ω. Thus, at 5 and 20 °C min−1 ω precipitates by diffusion at ~200 °C (Fig. 10a, b) at solute depleted regions of β [30, 50]. Diffusion-driven element partitioning in which alloying elements are rejected from ω particles is responsible for further growing of this phase as it has been shown experimentally in other β-metastable alloys [9, 51]. The connection between ω and β is corroborated in Fig. 11a, b by the crystallographic relationship between ω and β [46], i.e., \( a_{\omega } = a_{\beta } \sqrt 2 \), for the 5 and 20 °C min−1 at the onset of ω precipitation, indicating that ω grows from β crystals. On the other hand, at 50 °C min−1 heating rate (Fig. 11c) the a ω cell parameter starts with the last values obtained for c α″ before the athermal α″ → β reversion. However, the ω reflections start to be resolvable by diffraction after the α″ → β reversion (Fig. 8b). This suggests that ω forms at former α″ sites for 50 °C min−1. A similar correlation was also observed in pure titanium deformed under high pressures for a γ phase with the same orthorhombic symmetry as α″ [52, 53]. Further evidences of the formation of ω from α″ are reported in [35, 54, 55] for quenched metastable β titanium alloys. Therefore, the formation of ω at 50 °C min−1 may be induced during the last stages of the athermal α″ → β reversion by the complete and sudden reshearing of lattice strains shown experimentally in other metastable β alloys [9, 51]. Furthermore, the α″ phase is retained during heating for lower heating rates as described below, which may prevent the formation of high volume fraction of ω.
Stabilization of α″
The volume fraction of α″ for the 5 °C min−1 condition slowly increases as the temperature increases above the end of the athermal α″ → β reversion (~260 °C, see Table 4) up to ~400 °C (Fig. 10a). On the other hand, for the 20 °C min−1 condition the increase of the volume fraction of α″ is steeper until the same content (~45 vol%) as for 5 °C min−1 is achieved at 420 °C (Fig. 10b). These changes are directly related to the evolution of the cell parameters of α″ (Fig. 11a, b). Here, the 20 °C min−1 condition presents a steeper increase and decrease of the b α″ and a α″ cell parameters, respectively, in comparison with 5 °C min−1. At 400 °C, b α″ and a α″ coincide at both heating rates. The changes in the cell parameters are an indication of diffusion-driven element partitioning between phases and may be related to a transfer of β-stabilizer atoms (Fe and V) from α″ into β as it has been suggested for alloys with similar [Mo]eq concentrations [6, 56]. For this reason the phase observed within this temperature range is called \( \alpha^{\prime\prime}_{\text{lean}} \) hereafter. Therefore, the larger increase in the volume fraction of \( \alpha^{\prime\prime}_{\text{lean}} \) observed in the 20 °C min−1 condition up to 400 °C is a diffusive process. Simultaneously, the remaining β phase is chemically stabilized. As a result, the diffusionless α″ to β reversion caused by thermal stresses is partly suppressed for lower heating rates due to element partitioning. \( \alpha^{\prime\prime}_{\text{lean}} \) phase is retained for lower heating rates, which reduces the maximum attained volume fraction of ω phase.
In the same temperature region as described above, the volume fraction of ω reaches a maximum and subsequently decreases for all heating rates. At 5 °C min−1, this decrease follows practically the same trend as the increase in β (Fig. 10a) suggesting an ω → β transformation. The same trend is observed for 50 °C min−1 (Fig. 10c). This transformation is not evident for the 20 °C min−1 owing to the low concentration of ω and the previously explained β-stabilization. The changes for the 50 °C min−1 condition are also evidenced by a decrease of a ω at ~370 °C (Fig. 11c). Furthermore, as the temperature increases, a ω shifts to the position of c α″ in the ranges 340–380 °C, 370–400 °C, and 443–448 °C for 5, 20, and 50 °C min−1, respectively (Fig. 11a) to c). The same correlation of cell parameters was described in “Formation of ω” section and in [52, 53]. This, together with the continuous shift of {002}ω reflections to a position close to that of {130}α″ (see Online Resources 1, 2, and 3) also points to the formation of α″ from ω. A prior work [35] suggested that band-like products of ω transform into α″ within martensite plates during holding at ~450 °C for a Ti–8Mo wt% alloy. Furthermore, formation of α″ during isothermal decomposition of the β-metastable phase has been reported at moderate temperatures (420–600 °C) for several alloys (Ti-17 [12], Ti-5553 [57], VT-22, and TIMETAL-LCB [8] with [Mo]eq = ~9, 12, 11, 18 wt%, respectively). This product has been referred to as isothermal α″ since its formation was observed during heat treatments at constant temperature [8, 12, 57, 58]. The same nomenclature is used here (\( \alpha^{\prime\prime}_{\text{iso}} \)), but it must be taken into account that this serves only to distinguish between two different formation mechanisms of this orthorhombic phase: \( \alpha^{\prime\prime}_{\text{lean}} \) forms from β, while \( \alpha^{\prime\prime}_{\text{iso}} \) forms from ω. It is important to note that the y-coordinate of \( \alpha^{\prime\prime}_{\text{iso}} \) (as determined at 50 °C min−1) is slightly different (0.181) from the one obtained by the combination of \( \alpha^{\prime\prime}_{\text{iso}} \) and \( \alpha^{\prime\prime}_{\text{lean}} \) (5 and 20 °C min−1) suggesting compositional variations between them.
The transformations explained in this section correlate with the endothermic and exothermic peaks 3 and 4 of the DSC as shown in Table 4. Due to the higher magnitude of the β → \( \alpha^{\prime\prime}_{\text{lean}} \) transformation compared to the ω → \( \alpha^{\prime\prime}_{\text{iso}} \) transformation in the 20 °C min−1 condition, the overlapping of both processes is observed as a single endothermic peak in the DSC curve.
Formation of α
Transformation from the β phase
Reflections close to those expected for α are visible for the 50 °C min−1 condition from ~425 °C (Fig. 8). Furthermore, evidences of α reflections are marked by arrows in Fig. 9b, c for the 20 and 50 °C min−1 conditions, respectively. Also, Figs. 10 and 11 show that all heating rates undergo a decrease in the volume fraction of β and in the β cell parameter above ~390, ~400, and ~450 °C for the 5, 20, and 50 °C min−1 conditions, respectively. The presence of α at 400 °C has been reported during isothermal aging of Ti–10V–2Fe–3Al [59, 60]. Figure 2 and Table 4 show that the β → α transformation is reflected in the exothermic reaction number 4. At 5 °C min−1 it takes place as a single reaction between 415 and 435 °C, at 20 °C min−1 occurs between 416 and 450 °C, while at 50 °C min−1 overlaps with the ω → \( \alpha^{\prime\prime}_{\text{iso}} \) transformation (454–480 °C) as it has also been suggested for a Ti-LCB metastable β alloy [14]. At higher temperatures, an increase of the β content of ~10 vol% is observed for all conditions suggesting that an additional \( \alpha^{\prime\prime}_{{{\text{lean}}/{\text{iso}}}} \to \beta \) transformation takes place. The temperature ranges of this transformation fit with the endothermic peak 5 of the DSC (Table 4).
Evolution of α″
Figure 11 shows a progressive evolution of b α″ , c α″ , and a α″ towards \( a_{\alpha } \sqrt 3 \), c α and a α , respectively, until they coincide above temperatures between 555 and 660 °C. This transformation is associated with the exothermic peak number 6 of the DSC (Fig. 2; Table 4). This indicates a gradual transformation of the orthorhombic \( \alpha^{\prime\prime}_{{{\text{lean}}/{\text{iso}}}} \) into α. The formation of α from α″ has been proposed in [61], where homogeneous α precipitates were observed by TEM inside the stacking faults of martensitic α″ plates for quenched samples of a TC21 alloy aged at 500 °C. On the other hand, α was observed between \( \alpha^{\prime\prime}_{\text{iso}} \) for quenched samples of TIMETAL-LCB aged at 538 °C [8]. Moreover, the \( \alpha^{\prime\prime}_{\text{lean}} \to \alpha \) transformation was also suggested in the basis of dilatometry tests for the current alloy [6]. Figure 11 shows a direct evidence of this phase transformation. Figure 12a shows a TEM bright field image of an α″ martensite plate surrounded by a non-uniform distribution of fine α laths within the metastable β matrix for a 20 °C min−1 sample quenched from 540 °C. Moreover, α laths marked by arrows have formed within the martensite plate. The dark field TEM image in Fig. 12b shows that particles of α″ (bright regions) are also found dispersed in the metastable β matrix. At this temperature α″ already presents a symmetry close to hcp (Fig. 11b) and, therefore, these particles may correspond to particles undergoing the α″ → α transformation. As a consequence, the formation of α from α″ can occur by two different paths:

TEM of the 20 °C min−1 condition heated up to 540 °C and subsequently water quenched: a bright field image where an α″ martensite plate surrounded by a non-uniform distribution of fine α laths in the metastable β matrix can be observed, and b dark field image using the \( \left( {1\bar{1}\bar{1}} \right) \) α″ reflection showing fine α″ in the metastable β matrix
-
(1)
Formation of α from the α″ martensite plates, which corresponds to \( \alpha^{\prime\prime}_{\text{lean}} \) → α transformation.
-
(2)
Formation of α from \( \alpha^{\prime\prime}_{\text{iso}} \) particles distributed within the metastable β matrix previously formed from the ω → \( \alpha^{\prime\prime}_{\text{iso}} \) phase transformation.
The lattice correspondence between α″ and α can be established from the shift of α″ reflections to the positions of α, as shown in Fig. 7, and the correspondence between the evolution of the cell parameters shown in Fig. 11. These relationships are established as follows: \( \left\{ {130\} \alpha \prime \prime } \right. \parallel \left\{ {110} \right\} \alpha \) and \( \left\{ {020\} \alpha \prime \prime } \right. \parallel \left\{ {100} \right\} \alpha \). The hcp and orthorhombic structures are shown together in Fig. 13 as a projection along the c axis (a 3D animation is provided in Online Resource 4).

Representation of the lattice correspondence observed between the α″ and α phases. The atoms in the basal planes of α and α″ are indicated by yellow and red spheres, respectively. The atoms in the consecutive central plane of the α and α″ lattices are depicted by green and orange spheres, respectively (Color figure online)
Dissolution of α
The last endothermic peak of the DSC (number 7) corresponds to the transformation of α into β (Fig. 2; Table 4). A peak shift to higher temperatures can be observed as a function of the heating rate. This process finishes at temperatures close to the equilibrium β-transus temperature (800 °C) [20].
Conclusions
The phase transformation kinetics of the β quenched Ti–10V–2Fe–3Al alloy has been studied by in situ HEXRD and DSC as a function of heating rate. The following conclusions can be drawn from the investigations:
-
A fast athermal α″ → β reversion is observed at ~200 °C for all heating rates (5, 20, and 50 °C min−1). The magnitude of this athermal α″ → β reversion increases with the heating rate leading to a complete transformation of α″ into β for the 50 °C min−1 condition.
-
Two different origins for the formation of ω are identified depending on the heating rate: at 5 and 20 °C min−1 ω grows from β crystals by a diffusion controlled mechanism, while at 50 °C min−1 ω forms from the orthorhombic α″ phase. The latter may be induced during the last stages of the α″ → β reversion by the complete and sudden reshearing of lattice strains.
-
Two different mechanisms of formation of α″ are observed at temperatures above the end of the α″ → β reversion: on the one hand, \( \alpha^{\prime\prime}_{\text{lean}} \) grows to stabilize the excess of β formed during the martensite reversion (5 and 20 °C min−1), while at higher temperatures \( \alpha^{\prime\prime}_{\text{iso}} \) forms from ω as reflected by the corresponding evolution of the cell parameters.
-
At moderate temperatures (420–650 °C) α forms through three different paths:
-
(a)
Formation of α from the α″ martensite plates, which corresponds to \( \alpha^{\prime\prime}_{\text{lean}} \) → α transformation.
-
(b)
Formation of α from \( \alpha^{\prime\prime}_{\text{iso}} \) particles distributed within the metastable β matrix previously formed from the ω → \( \alpha^{\prime\prime}_{\text{iso}} \) phase transformation.
-
(c)
Formation of α from the β phase (β → α).
-
(a)
-
In situ HEXRD has been applied to follow univocally the evolution of the single Bragg reflections of each phase allowing a complete determination of the phase transformation sequence in the Ti–10V–2Fe–3Al metastable β alloy as a function of heating rate.
References
Banerjee D, Williams JC (2013) Perspectives on titanium science and technology. Acta Mater 61:844–879
Boyer RR, Briggs RD (2005) The use of titanium alloys in the aerospace industry. J Mater Eng Perform 14:681–685
Maeshima T, Nishida M (2004) Shape memory and mechanical properties of biomedical Ti–Sc–Mo alloys. Mater Trans 45:1101–1105
Duerig TW, Williams JC (1984) Microstructure and properties of beta titanium alloys. In: Boyer RR, Rosenberg HW (eds) Beta titanium alloys in the 1980’s. The Metallurgical Society of AIME, Atlanta, pp 16–69
Miyazaki S, Sachdeva RL (2009) Shape memory effect and superelasticity in Ti–Ni alloys. In: Yoneyama T, Miyazaki S (eds) Shape memory alloys for biomedical applications. Woodhead, Cambridge, pp 3–18
Duerig TW, Albrecht J, Richter D, Fischer P (1982) Formation and reversion of stress induced martensite in Ti–10V–2Fe–3Al. Acta Metall 30:2161–2172
Ohmori Y, Ogo T, Nakai K, Kobayashi S (2001) Effects of ω-phase precipitation on β → α, α″ transformations in a metastable β titanium alloy. Mater Sci Eng A 312:182–188
Ivasishin OM, Markovsky PE, Semiatin SL, Ward CH (2005) Aging of coarse- and fine-grained β titanium alloys. Mater Sci Eng A 405:296–305
Nag S, Banerjee R, Srinivasan R, Hwang JY, Harper M, Fraser HL (2009) ω-Assisted nucleation and growth of α precipitates in the Ti–5Al–5Mo–5V–3Cr–0.5Fe β titanium alloy. Acta Mater 57:2136–2147
Bein S, Béchet J (1996) Phase transformation kinetics and mechanisms in titanium alloys Ti-6.2.4.6, β-CEZ and Ti-10.2.3. J Phys IV 6:99–108
Malinov S, Sha W, Markovsky P (2003) Experimental study and computer modeling of the β → α+β phase transformation in β21s alloy at isothermal conditions. J Alloys Compd 348:110–118
Bruneseaux F, Geandier G, Gautier E, Appolaire B, Dehmas M, Boulet P (2007) In situ characterization of the transformation sequences of Ti17 alloy by high energy X-ray diffraction: Influence of the thermal path. In: Niinomi M, Akiyama S, Hagiwara M, Ikeda M, Maruyama K (eds) Ti-2007 science and technology. The Japan Institute of Metals, Sendai, pp 563–566
Contrepois Q, Carton M, Lecomte-Beckers J (2011) Characterization of the β phase decomposition in Ti–5Al–5Mo–5V–3Cr at slow heating rates. Open J Met 1:1–11
Carton M, Jacques P, Clément N, Lecomte-Beckers J (2007) Study of transformations and microstructural modifications in Ti-LCB and Ti-555 alloys using differential scanning calorimetry. In: Niinomi M, Akiyama S, Hagiwara M, Ikeda M, Maruyama K (eds) Ti-2007 science and technology. The Japan Institute of Metals, Sendai, pp 491–494
Liss KD, Bartels A, Schreyer A, Clemens H (2003) High-energy X-rays: a tool for advanced bulk investigations in materials science and physics. Texture Microstruct 35:219–252
Aeby-Gautier E, Bruneseaux F, Teixeira J, Appolaire B, Geandier G, Denis S (2007) Microstructural formation in Ti alloys: in-situ characterization of phase transformation kinetics. JOM 59:54–58
Jones NG, Dashwood RJ, Jackson M, Dye D (2009) β phase decomposition in Ti–5Al–5Mo–5V–3Cr. Acta Mater 57:3830–3839
Wollmann M, Kiese J, Wagner L (2011) Properties and applications of titanium alloys in transport. In: Zhou L, Chang H, Lu Y, Xu D (eds) Ti-2011. Science Press Beijing, Beijing, pp 837–844
Eylon D, Vassel A, Combres Y, Boyer RR, Bania PJ, Schutz RW (1994) Issues in the development of beta titanium alloys. JOM 46:14–15
Boyer R, Welsch G, Collings EW (1994) Material properties handbook: titanium alloys, 4th edn. ASM International, Ohio
Lin W, Dalmazzone D, Fürst W, Delahaye A, Fournaison L, Clain P (2013) Acurate DSC measurement of the phase transition temperature in the TBPB–water system. J Chem Thermodyn 61:132–137
Pompe G, Schulze U, Hu J, Pionteck J, Höhne GWH (1999) Separation of glass transition and melting polyethylene/poly(butyl-methacrylate-co-methyl-methacrylate) interpenetrating polymer networks in TMDSC and DSC curves. Thermochim Acta 337:179–186
ID15 - High Energy Scattering Beamline, ESRF (1999). http://www.esrf.eu/UsersAndScience/Experiments/StructMaterials/ID15. Accessed 09 July 2014
P07 - The High Energy Materials Science Beamline, DESY (2010). http://photon-science.desy.de/facilities/petra_iii/beamlines/p07_high_energy_materials_science/index_eng.html. Accessed 09 July 2014
Staron P, Fischer T, Lippmann T, Stark A, Daneshpour S, Schnubel D, Uhlmann E, Gerstenberger R, Camin B, Reimers W, Eidenberger E, Clemens H, Huber N, Schreyer A (2011) In situ experiments with synchrotron high-energy X-rays and neutrons. Adv Eng Mater 13:658–663
Hammersley A, Svensson SO, Thompson A (1994) Calibration and correction of spatial distortions in 2D detector systems. Nucl Instrum Methods Phys Res A 346:312–321
Abramoff MD, Magalhães PJ, Ram SJ (2004) Image processing with imageJ. Biophoton Int 11:36–41
Lutterotti L, Matthies S, Wenk HR, Schultz AJ, Richardson J (1997) Combined texture and structure analysis of deformed limestone from time-of-flight neutron diffraction spectra. J Appl Phys 81:594–600
Cai MH, Lee CY, Kang S, Lee YK (2011) Fine-grained structure fabricated by strain-induced martensite and its reverse transformations in a metastable β titanium alloy. Scripta Mater 64:1098–1101
Banerjee S, Mukhopadhyay P (2007) Phase transformations: examples from titanium and zirconium alloys, 1st edn. Elsevier, Oxford
Willams JC, Fontaine D, Paton NE (1973) The ω-phase as an example of an unusual shear transformation. Metall Trans 4:2701–2708
Sass SL (1972) The structure and decomposition of Zr and Ti B.C.C. solid solutions. J Less Common Met 28:157–173
Li SJ, Cui TC, Hao YL, Yang R (2008) Fatigue properties of a metastable β-type titanium alloy with reversible phase transformation. Acta Biomater 4:305–317
Dutta J, Ananthakrishna G, Banerjee S (2012) On the athermal nature of the β to ω transformation. Acta Mater 60:556–564
Mantani Y, Takemoto Y, Hida M, Sakakibara A, Tajima M (2004) Phase transformation of a α″ martensite structure by aging in Ti-8 mass%Mo alloy. Mater Trans 45:1629–1634
Popov AA, Illarionov AG, Grib SV, Elkina AO (2011) Formation conditions of omega phase in the titanium alloys after quenching. In: Zhou L, Chang H, Lu Y, Xu D (eds) Ti-2011. Science Press Beijing, Beijing, pp 694–697
Bönisch M, Calin M, Waitz T, Panigrahi A, Zehetbauer M, Gebert A, Skrotzki W, Eckert J (2013) Thermal stability and phase transformations of martensitic Ti–Nb alloys. Sci Technol Adv Mater 14:1–9
Liss KD, Stark A, Bartels A, Clemens H, Buslaps T, Phelan D, Yeoh LA (2008) Directional atomic rearrangements during transformations between the α- and γ-phases in titanium aluminides. Adv Eng Mater 10:389–392
Ramsteiner IB, Shchyglo O, Mezger M, Udyansky A, Bugaev V, Schöder S, Reichert H, Dosch H (2008) Omega-like diffuse X-ray scattering in Ti–V caused by static lattice distortions. Acta Mater 56:1298–1305
Settefrati A, Dehmas M, Geandier G, Denand B, Aeby-Gautier E, Appolaire B, Khelifati G, Delfosse J (2011) Precipitation sequences in beta metastable phase of Ti-5553 alloy during ageing. In: Zhou L, Chang H, Lu Y, Xu D (eds) Ti-2011. Science Press Beijing, Beijing, pp 468–472
Basak CB, Neogy S, Srivastava D, Dey GK, Banerjee S (2011) Disordered bcc γ-phase to δ-phase transformation in Zr-rich U-Zr alloy. Philos Mag 91:3290–3306
Banerjee D, Muraleedharan K, Strudel JL (1998) Substructure in titanium alloy martensite. Philos Mag A 77:299–323
Obbard EG, Hao YL, Talling RJ, Li SJ, Zhang YW, Dye D, Yang R (2011) The effect of oxygen on α″ martensite and superelasticity in Ti–24Nb–4Zr–8Sn. Acta Mater 59:112–125
Banumathy S, Mandal RK, Singh AK (2009) Structure of orthorhombic martensitic phase in binary Ti–Nb alloys. J Appl Phys 106:093518.1–093518.6
Brown ARG, Clark D, Eastabrook J, Jepson KS (1964) The titanium–niobium system. Nature 201:914–915
Aurelio G, Fernández-Guillermet A, Cuello GJ, Campo J (2002) Metastable phases in the Ti–V system: part I. Neutron diffraction study and assessment of structural properties. Metall Mater Trans A 33:1307–1317
Kim HY, Ikehara Y, Kim JI, Hosoda H, Miyazaki S (2006) Martensitic transformation, shape memory effect and superelasticity of Ti–Nb binary alloys. Acta Mater 54:2419–2429
Williams JC, Hickman BS, Marcus HL (1971) The effect of omega phase on the mechanical properties of titanium alloys. Metall Trans 2:1913–1919
Šmilauerová J, Janeček M, Harcuba P, Stráský J (2013) Aging study of TIMETAL LCB titanium alloy. Proc. Met.. http://www.metal2014.com/files/proceedings/12/reports/1536.pdf. Accessed 09 July 2014
Hickman BS (1969) The formation of omega phase in titanium and zirconium alloys: a review. J Mater Sci 4:554–563. doi:10.1007/BF00550217
Devaraj A, Nag S, Srinivasan R, Williams REA, Banerjee S, Banerjee R, Fraser HL (2012) Experimental evidence of concurrent compositional and structural instabilities leading to ω precipitation in titanium–molybdenum alloys. Acta Mater 60:596–609
Akahama Y, Kawamura H, Bihan TL (2001) New δ (distorted-bcc) titanium to 220 GPa. Phys Rev Lett 87:275503.1–275503.4
Vohra YK, Spencer PT (2001) Novel gamma-phase of titanium metal at megabar pressures. Phys Rev Lett 86:3068–3071
Duerig TW, Middleton RM, Terlinde GT, Williams JC (1980) Stress assisted transformation in Ti–10V–2Fe–3Al. In: Kimura H, Izuma O (eds) Titanium ‘80 science and technology. Metallurgical Society of AIME, Kyoto, pp 1503–1508
Davis R, Flower HM, West DRF (1979) Martensitic transformations in Ti–Mo alloys. J Mater Sci 14:712–722. doi:10.1007/BF00772735
Bagariatskii IA, Nosova GI, Tagunova TV (1958) Factors in the formation of metastable phases in titanium-base alloys. Sov Phys Dokl 3:1014–1018
Settefrati A, Aeby-Gautier E, Appolaire B, Dehmas M, Geandier G, Khelifati G (2013) Low temperature transformation in the β-metastable Ti-5553 alloy. Mater Sci Forum 738–739:97–102
Malinov S, Sha W, Guo Z, Tang CC, Long AE (2002) Synchrotron X-ray diffraction study of the phase transformations in titanium alloys. Mater Charact 48:279–295
Duerig TW, Terlinde GT, Williams JC (1980) Phase transformations and tensile properties of Ti–10V–2Fe–3Al. Metall Trans A 11:1987–1998
Ohmori Y, Natsui H, Nakai K, Ohtsubo H (1998) Effects of ω phase formation on decomposition of α″/β duplex phase structure in a metastable β Ti alloy. Mater Trans JIM 39:40–48
Tang B, Kou HC, Wang YH, Zhu ZS, Zhang FS, Li JS (2012) Kinetics of orthorhombic martensite decomposition in TC21 alloy under isothermal conditions. J Mater Sci 47:521–529. doi:10.1007/s10853-011-5829-5
Acknowledgements
The authors acknowledge the COMET-Program of the Austrian Research Promotion Agency (FFG) and the Province of Upper Austria (LOÖ), Grant No. 820492 for financial support. The European Synchrotron Radiation Facility (ESRF) and the Deutsches Elektronen-Synchrotron (DESY) are acknowledged for the provision of synchrotron radiation facilities in the framework of the MA1268 and I-20100329 EC proposals, respectively. The University Service for Transmission Electron Microscopy (USTEM) of the Vienna University of Technology is acknowledged for the provision of the transmission electronic microscope. Böhler Schmiedetechnik GmbH & Co KG is also acknowledged for the provision materials. The authors would like to thank Dr. A. Rhys Williams for proof-reading the manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Online Resource 1: Video sequence of a color-coded 2D plot corresponding to a selected portion of complete Debye–Scherrer rings converted into Cartesian coordinates. The evolution of the Bragg reflections is shown as a function of temperature during heating at 5 °C min-1 (MPG 8174 kb)
Online Resource 2: Video sequence of a color-coded 2D plot corresponding to a selected portion of complete Debye–Scherrer rings converted into Cartesian coordinates. The evolution of the Bragg reflections is shown as a function of temperature during heating at 20 °C min-1 (MPG 4964 kb)
Online Resource 3: Video sequence of a color-coded 2D plot corresponding to a selected portion of complete Debye–Scherrer rings converted into Cartesian coordinates. The evolution of the Bragg reflections is shown as a function of temperature during heating at 50 °C min-1 (MPG 10122 kb)
Online Resource 4: 3D animation of the lattice correspondence observed between the a? and a phases (MPG 17818 kb)
Rights and permissions
About this article

Cite this article
Barriobero-Vila, P., Requena, G., Warchomicka, F. et al. Phase transformation kinetics during continuous heating of a β-quenched Ti–10V–2Fe–3Al alloy. J Mater Sci 50, 1412–1426 (2015). https://doi.org/10.1007/s10853-014-8701-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-014-8701-6