
Abstract
Nanoscale SrHPO4 is prepared via a polyol-mediated synthesis. The resulting particles are well crystallized, non-agglomerated, and very uniform in size and shape. By adjusting the experimental conditions, SrHPO4 can be obtained with the α-type as well as with the β-type of modification. In particular, particle diameters of 16 nm (α-SrHPO4) and 12 nm (β-SrHPO4) are obtained. The title compound is characterized by scanning electron microscopy, dynamic light scattering, X-ray powder diffraction, and infrared spectroscopy.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
SrHPO4 has been claimed as a catalyst, proton conductor, and surface conditioner as well as for application in batteries, fuel cells, for flame proofing, and thermal cathodes [1–4]. Aiming at catalysis, the use of SrHPO4 has been described for amination reactions, in particular, for its outstanding selectivity and conversion rate in the synthesis of diazabicyclo[2,2,2]octane [1]. Crystallograpically, SrHPO4 has been described with three different types of modifications: α-SrHPO4 is characterized by two crystallographically independent strontium sites—both with eightfold coordination to form a distorted quadratic antiprism [5]. Surprisingly, little is known with concern to β-SrHPO4 as the thermodynamically most stable modification. Here, even sufficient single crystal structure data are still lacking. Finally, γ-SrHPO4 has been reported to contain two independent Sr sites, which are both coordinated by nine oxygen atoms [6].
With concern to all applications mentioned above, a straight-forward access to nanoscale SrHPO4 exhibiting non-agglomerated particles with high surface area can be of importance for fundamental as well as for applicational issues. Moreover, the question regarding crystallinity and type of modification of the as-prepared nanomaterial as well as the possibility to steer the type of modification arises. Surprisingly, the synthesis of nanoscale hydrogen phosphates as well as phosphates of the alkaline earth metals has been barely tackled, in general [7, 8]. In detail, SrHPO4 has been realized via a water based coprecipitation [3, 9]. To this end, belt-type morphologies, 50–100 nm in widths and several microns in lengths as well as agglomerated mesoscaled particles, 150–200 nm in diameter have been obtained. In this study, the synthesis of highly crystalline and non-agglomerated SrHPO4 with diameters below 50 nm is addressed. Furthermore, it is intended to steer the type of the crystallographic modification selectively by suited variation of the experimental conditions.
Experimental section
Synthesis
α-SrHPO4 was prepared in a typical recipe by dissolving Sr(NO3)2 (151 mg/0.71 mmol, >99%, Riedel-de Haën) in 1 mL of deionised water and mixing with 50 mL of diethylene glycol (DEG) in a 1,000 mL beaker (solution A). In addition, H3PO4 (68 mg/0.69 mmol, >99.999%, Sigma Aldrich) was dissolved in 1 mL of H2O and 5 mL of DEG (solution B). Solution A was heated to 170 °C and solution B rapidly added with vigorous stirring. By this measure an immediate particle nucleation was initiated. The resulting suspension was immediately quenched to 15 °C by addition of 150 mL of an ethanol/dry ice mixture. To separate the nanoparticles, the suspension was centrifuged (20 min, 26,000 rpm). The colorless nanomaterial was resuspended in and centrifuged from ethanol twice in order to remove all DEG and remaining salts. Finally, the powder was dried in a drying oven (15 min, 75 °C). The nanoscaled solid can be easily redispersed in DEG by sonification.
In a typical recipe for the polyol-mediated synthesis of nanoscale β-SrHPO4, 50 mL of DEG (99%, Acros) were placed in a three-neck flask (volume 100 mL) with a Claisen stillhead. Sr(NO3)2 (150 mg/0.71 mmol, >99%, Riedel-de Haën) was dissolved in 1 mL of deionised water and added to DEG (solution A). In addition, H3PO4 (68 mg/0.69 mmol, >99.999%, Sigma Aldrich) was dissolved in 5 mL of DEG (solution B). The synthesis was performed under a continuous nitrogen flow. Solution A was heated to 90 °C to dissolve all starting materials and then cooled to 75 °C. For optimal conditions of nucleation and growth, solution B was rapidly added at this temperature under vigorous stirring. After 5 min of stirring, the suspension was rapidly heated (within 5 min) to 190 °C. Then the suspension was left to cool to room temperature and diluted with 50 mL of ethanol. Finally, the colorless nanomaterial was washed and dried as described above.
Analytical characterization
Dynamic light scattering (DLS) was conducted with polystyrene cuvettes applying a Nanosizer ZS (Malvern Instruments). Samples were measured after resuspension in DEG. Scanning electron microscopy (SEM) was carried out with a Zeiss Supra 40 VP, equipped with a field-emission gun (samples deposited on silicon and sputtered with Pt; acceleration voltage 10 kV; working distance 3 mm). X-ray powder diffraction (XRD) analysis was conducted with a Stoe Stadi P system, using Ge-monochromatized Cu-Kα radiation. Fourier-transform infrared (FT-IR) spectra were recorded with a Bruker Vertex 70 FT-IR spectrometer; the samples were measured as pellets in KBr with a resolution of 4 cm−1.
Results and discussion
To realize SrHPO4 on the nanoscale, a polyol-mediated synthesis was applied. This type of synthesis has been widely used already to realize various nanomaterials, including elemental metals, oxides, sulfides or fluorides [10–12]. The background of synthesis, in general, is a multidentate and high-boiling alcohol (so-called polyol, e.g. diethylene glycol, ethylene glycol, glycerine) as the liquid phase. Due to the elevated temperature of synthesis (typically 150–300 °C) crystalline materials are often gained. The polyol furthermore coordinates, and therefore stabilizes the particle surface, which allows an efficient control of particle growth and agglomeration processes. Aiming at SrHPO4, DEG was used as the polyol, Sr(NO3)2 and H3PO4 were selected as the starting materials. For optimal conditions of nucleation and growth [13], precipitation of the title compound was carried out by mixing homogeneous solutions of the starting materials.
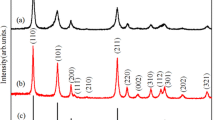
With regard to the crystallographic modification—by means of thermodynamics—β-SrHPO4 has been reported to represent the most stable modification at ambient conditions [5, 6]. Consequently, with increasing temperature and reaction time the formation of β-SrHPO4 should be favored. On the other hand, the formation of a metastable modification is expected under kinetic control of the reaction. This expectation is consistent with findings for the bulk compound. According to Mooney et al. crystallization of α-SrHPO4 has been observed as the initial product of chemical synthesis. Recrystallization of the thermodynamically stable β-form, thereafter is time-dependent and sometimes observed to occur spontaneously. In addition, transformation can be significantly fostered by increased temperatures [14, 15]. Analogous measures were evaluated here when aiming at nanoscale SrHPO4. Indeed, α-SrHPO4 was obtained according to the hot-injection method by mixing the starting materials at 170 °C. To suppress recrystallization, the suspension was immediately quenched by addition of an ethanol/dry ice mixture. Aiming at the β-modification, optimal conditions of nucleation mixing of the reactants were performed at moderate temperatures (75 °C). The resulting precipitate thereafter was crystallized by heating up to 190 °C. In accordance with Ostwald’s step rule [16], the α-modification was preserved by quenching, but can be recrystallized by prolonged heating. X-ray diffraction patterns of both as-prepared nanomaterials indicate β-SrHPO4 to be phase pure, and α-SrHPO4 to contain only minor amounts of the β-phase (Fig. 1) [17, 18].

XRD pattern of as-prepared α-SrHPO4 (top, reference: ICDD-No. 01-070-1215) and β-SrHPO4 (bottom, reference: ICDD-No. 012-0368)
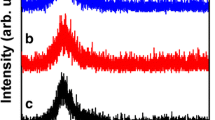
FT-IR spectroscopy was performed for further chemical characterization (Fig. 2). Spectra of α-SrHPO4 and β-SrHPO4 are almost identical, and also in accordance to those of bulk samples as published by Mel’nikova et al. [19]. To this end, the strong and broad band at 3,450 cm−1 is assigned to ν(O–H) [20]. Furthermore, three broad but weak bands between 3,000 and 1,500 cm−1 can be attributed to hydrogen bridge bonds. The manifold of sharp and partly splitted bands below 1,500 cm−1, finally can be related to ν(PO4), δ(PO4) as well as to δ(O–H) vibrations. The number and splitting of these vibrational bands are in accordance with the low site symmetry (1) of the [PO4]3− anion in the crystalline lattice [5].

FT-IR spectra of as-prepared α-SrHPO4 (bold) and β-SrHPO4 (dotted)
Particle size and size distribution of as-prepared α-SrHPO4 and β-SrHPO4 were verified based on electron microscopy (Fig. 3). Here, uniform particles with diameters of 16(3) nm (α-SrHPO4) and 12(2) nm (β-SrHPO4) are observed. These values were deduced based on a statistical evaluation of about 1,000 particles in each case. In addition to powder samples, as-prepared suspensions in DEG were investigated with DLS measurements (Fig. 4). With the values of 13(2) nm (α-SrHPO4) and 15(2) nm (β-SrHPO4), the resulting hydrodynamic radii confirm the findings of electron microscopy. Moreover, the concurrence of particle diameters steming from SEM and DLS as well as the very narrow size distribution strike on the uniformity of the as-prepared nanomaterials and the absence of agglomerates.

SEM images of as-prepared α-SrHPO4 (top) and β-SrHPO4 (bottom)

Particle size distribution of as-prepared nanoscale α-SrHPO4 (top) and β-SrHPO4 (bottom) in DEG suspension
Conclusions
Nanoscale SrHPO4 was prepared via a polyol-mediated synthesis. By specific selection of the experimental conditions the title compound was yielded with two different crystallographic modifications. Based on a hot-injection of precursors and a rapid quenching of the resulting suspension, the formation of α-SrHPO4 was initiated. While applying a continuous and slow heating process, the thermodynamically most stable β-SrHPO4 was gained. According to the analytical characterization (XRD, FT-IR, SEM, DLS) both the as-prepared nanomaterials turned out to be highly crystalline, non-agglomerated, and very uniform in size and shape. In particular, particle diameters of 16 nm (α-SrHPO4) and 12 nm (β-SrHPO4) were obtained.
References
Fischer A, Mallat T, Baiker A (1997) Catal Today 37:167. doi:https://doi.org/10.1016/S0920-5861(97)00009-6
Louati B, Guidara K, Gargouri M, Fourati M (2005) Z Naturforsch 60a:121
Kim J, Noh M, Cho J, Kim HM, Kim KB (2005) J Electrochem Soc 152:A1142. doi:https://doi.org/10.1149/1.1896526zz
Levchik SV, Weil ED (2006) J Fire Sci 24(5):364. doi:https://doi.org/10.1177/0734904106068426
Boudjada A, Masse R, Guitel JC (1978) Acta Crystallogr B 34:2692. doi:https://doi.org/10.1107/S0567740878009036
Taher LB, Smiri L, Laligant Y, Maisonneuve V (2000) J Solid State Chem 152:428. doi:https://doi.org/10.1006/jssc.2000.8700
Purnendu P, Ray AR, Ramanan A (2007) J Am Ceram Soc 90(4):1237. doi:https://doi.org/10.1111/j.1551-2916.2007.01508.x
Purnendu P, Ramanan A, Ray AR (2006) Am J Biochem. Biotechnol 2(2):61
Zheng Y, Cheng Y, Wang Y, Yu Y, Chen D, Bao F (2005) J Cryst Growth 280:569. doi:https://doi.org/10.1016/j.jcrysgro.2005.03.067
Toneguzzo P, Viau G, Acher O, Guillet F, Bruneton E, Fievet F (2000) J Mater Sci 35:3767. doi:https://doi.org/10.1023/A:1004864927169
Feldmann C, Jungk HO (2001) Angew Chem Int Ed 40:359. doi:10.1002/1521-3773(20010119)40:2<359::AID-ANIE359>3.0.CO;2-B
Feldmann C, Roming M, Trampert K (2006) Small 2:1248. doi:https://doi.org/10.1002/smll.200600140
LaMer VK, Dinegar RH (1950) J Am Chem Soc 72:4847. doi:https://doi.org/10.1021/ja01167a001
Aia MA, Mathers JE, Mooney RW (1964) J Chem Eng Data 9:335. doi:https://doi.org/10.1021/je60022a006
Mooney RW, Aia MA, Hoffman CWW, Ropp RC (1959) J Am Chem Soc 81:827. doi:https://doi.org/10.1021/ja01513a020
Ostwald W (1897) Z Phys Chem 22:289
Ropp RC, Aia MA, Hoffman CWW, Veleker TJ, Mooney RW (1959) Anal Chem 31:1163. doi:https://doi.org/10.1021/ac60151a026
Boudjada A, Masse R, Guitel JC (1978) Acta Crystallogr B 34:2692. doi:https://doi.org/10.1107/S0567740878009036
Mel’nikova RY, Dzyuba ED, Pechkovskii VV, Barannikova TI, Kovalishina VI (1982) Russ J Inorg Chem 27:1724
Weidlein J, Müller U, Dehnicke K (1988) Schwingungsspektroskopie. Thieme Verlag, Stuttgart
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Roming, M., Feldmann, C. Selective synthesis of α- and β-SrHPO4 nanoparticles. J Mater Sci 43, 5504–5507 (2008). https://doi.org/10.1007/s10853-008-2830-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-008-2830-8