
Abstract
The widely used piezoelectric Pb(Zr1−xTix)O3 ceramics have been known to have Zr4+ and Ti4+ randomly distributed on the B-site lattice in the ABO3 perovskite structure. In this study, we attempted to develop long range 1:1 B-site cation order by forming the solid solution of (1 − x)Pb(Mg1/2W1/2)O3 − xPb(Zr0.5Ti0.5)O3 (x ≥ 0.60). High temperature X-ray diffraction tests indicate that the cation order is embedded in the structural order. The solid solution ceramics appear to have a non-cubic paraelectric phase above their Curie temperatures. The competition between the antiferroelectric order in Pb(Mg1/2W1/2)O3 and the ferroelectric order in Pb(Zr0.5Ti0.5)O3 leads to the relaxor ferroelectric behavior in the solid solution. Since the temperature at dielectric maximum, T m, is significantly above room temperature, regular polarization versus electric field hysteresis loops are recorded in these compositions at room temperature. In addition, these ceramics show very good piezoelectric properties.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Complex perovskite oxides usually contain two cation species with different charges on the B-site of the ABO3 structure [1]. Most Pb-containing complex perovskite oxides are ferroelectric, such as Pb(Mg1/3Nb2/3)O3 [2–6], Pb(Sc1/2Ta1/2)O3 [7], and Pb(Mg1/2W1/2)O3 [8–15]. Due to their unique dielectric and ferroelectric properties, these compounds have found wide applications in capacitors, actuators, transducers, resonators, and sensors. When a large difference in size (elastic energy) and charge (electrostatic energy) exists between B-site cations, long range cation order may develop in complex perovskites. This is the case in Pb(Mg1/2W1/2)O3, where Mg2+ (0.72 Å) and W6+ (0.60 Å) occupy different B-site sublattices (denoted as B′ and B′′ in literature) to form the double perovskite structure [8–12]. In Pb(Sc1/2Ta1/2)O3, Sc3+ (0.745 Å) and Ta5+ (0.64 Å) have moderate differences in both size and charge. As a result, the formation of long range 1:1 cation order requires an extended high temperature annealing process [7]. In the case of Pb(Mg1/3Nb2/3)O3, the B-site cation ordered domains are limited to the nanometer scale (<5 nm) and do not grow under any heat treatment [4–6].
The degree of B-site cation order strongly influences the dielectric and ferroelectric properties of complex perovskites. Setter and Cross [7] found that ordered Pb(Sc1/2Ta1/2)O3 crystals showed a sharp ferroelectric transition, while disordered crystals displayed a diffuse ferroelectric transition with a strong frequency dispersion. The weak cation order in Pb(Mg1/3Nb2/3)O3 can be significantly enhanced by chemical modification [4–6]. The direct correlation between the B-site cation order and the electric dipole order has been observed in Pb(Mg1/3Nb2/3)O3-based ceramics [4–6], as well as Pb(Mg1/3Ta2/3)O3-based and Pb(Sc2/3W1/3)O3-based ceramics [16, 17].
Pb(Zr1−xTix)O3, a solid solution between PbZrO3 and PbTiO3, has been the oxide system dominating the piezoelectric ceramics market for decades. The best piezoelectric properties are found in compositions close to the morphotropic phase boundary (MPB), roughly Pb(Zr0.5Ti0.5)O3 [18]. Even though a large size difference exists between Zr4+ (0.72 Å) and Ti4+ (0.605 Å), random occupation on the B-site lattice is observed due to the lack of charge difference. It would be of great interest to develop and manipulate cation order in the Pb(Zr0.5Ti0.5)O3 oxide, which could provide an additional variable to further improve the electromechanical properties.
In order to develop cation order in Pb(Zr0.5Ti0.5)O3, a charge difference needs to be introduced. For this purpose, we propose to study the (1 − x)Pb(Mg1/2W1/2)O3 − xPb(Zr0.5Ti0.5)O3 pseudo-binary solid solution system. First of all, a large charge difference as well as a large size difference exists between Mg2+ (0.72 Å) and W6+ (0.60 Å) [19]. Secondly, Mg2+ (0.72 Å) is the same size as Zr4+ (0.72 Å) and W6+ (0.60 Å) is similar in size to Ti4+ (0.605 Å) [19]. The size difference will maintain constant, while the charge difference will vary continuously with composition x in this system. Therefore, the contribution of charge difference to cation order can be isolated from that of size difference. Finally, Pb(Mg1/2W1/2)O3 is an antiferroeletic double perovskite with an orthorhombic distortion [13–15], while Pb(Zr0.5Ti0.5)O3 is a normal ferroelectric with a tetragonal distortion [18]. An MPB is expected in the solid solution, and it is expected that compositions near this phase boundary will exhibit peculiar properties.
Experimental
In order to ensure chemical homogeneity, perovskite powders in the (1 − x)Pb(Mg1/2W1/2)O3 − xPb(Zr0.5Ti0.5)O3 (x ≥ 0.60) pseudo-binary solid solution system were prepared by mixing the B-site oxides first. High purity (better than 99.9 wt.%) powders of MgO, WO3, ZrO2, and TiO2 were milled for 3 h using magnesia-stabilized zirconia mill media with isopropanol alcohol. The B-site oxide mixture was calcined at 1,100 °C for 4 h. Then, PbO with 2 at.% excess was added to the calcined powder. After milling for 3 h, the powder was calcined at 900 °C for 4 h to form the perovskite phase. Next, the powder was milled again for 2 h before sintering. Pellets were formed by uniaxial pressing with binder. Sintering was carried out between 1,150 and 1,250 °C for 3 h followed by a slow cooling at 8 °C/h. In order to minimize the Pb loss during sintering, a double crucible configuration with protective powders was used.
After sintering, the surface layers were removed by mechanical grinding. The density of the ceramics was measured using the Archimedes method. X-ray diffraction analysis was performed with Cu Kα radiation to determine phase purity and degree of ordering on the B-site. For the ceramic of composition x = 0.6, i.e., 0.40Pb(Mg1/2W1/2)O3 − 0.60Pb(Zr0.5Ti0.5)O3, X-ray diffraction was further performed at a series of temperatures up to 500 °C. After Au film electrodes were applied to the sample, the ceramic pellets were heated from room temperature to 350 °C at 2 °C/min for the dielectric property measurement. The ferroelectric polarization versus field hysteresis loop was recorded at room temperature at a series of electric fields. For piezoelectric property measurement, rectangular specimens with dimensions of 1.5 mm × 2.0 mm × 3.0 mm were cut from the sintered ceramics. Gold film electrodes were deposited on the 1.5 mm × 2.0 mm faces and poling was performed at 140 °C under 25 kV/cm. The piezoelectric coefficient was measured with a quasi-static d 33 meter.
Results and discussion
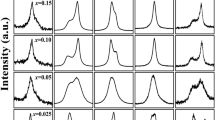
X-ray diffraction confirmed that a pure perovskite phase was achieved in all the prepared compositions of (1 − x)Pb(Mg1/2W1/2)O3 − xPb(Zr0.5Ti0.5)O3, as indicated in Fig. 1. Furthermore, in addition to the primary peaks of perovskite structure, the (½ ½ ½) superlattice peak, as shown in the inset, starts to emerge in the composition of x = 0.8 and gets stronger as x decreases. The presence of the (½ ½ ½) superlattice peak has been used to evaluate the degree of cation order in Pb(Mg1/3Nb2/3)O3-based complex perovskite since its theoretical intensity is weakly related to composition [4–6]. However, the situation is more complex in the present (1 − x)Pb(Mg1/2W1/2)O3 − xPb(Zr0.5Ti0.5)O3 pseudobinary system. Presumably the 1:1 B-site cation order takes the form of Pb[(Mg(1−x)Zrx)1/2(W(1−x)Tix)1/2]O3 based on their ionic sizes, i.e., Mg2+ and Zr4+ share the B′-sublattice while W6+ and Ti4+ share the B″-sublattice. Since the atomic scattering factor for X-ray diffraction scales with the atomic number (Z Mg = 12, Z Zr = 40, Z Ti = 22, Z W = 74), the theoretical intensity of the (½ ½ ½) superlattice peak is directly determined by the difference in the effective atomic number between the B′- and the B″-sublattices. The calculated theoretical intensity of the (½ ½ ½) peak based on the 1:1 cation order model is shown in Fig. 2, assuming an Fm3m symmetry. It is interesting to notice the theoretical intensity varies dramatically with the composition x. Furthermore, zero intensity is expected at x ≈ 0.75. That is to say, the (½ ½ ½) superlattice shall not appear even in a fully ordered ceramic with this composition. Therefore, in the (1 − x)Pb(Mg1/2W1/2)O3 − xPb(Zr0.5Ti0.5)O3 system, the superlattice intensity may not be used to evaluate the degree of the 1:1 cation order. The monotonic increase in the superlattice intensity with decreasing x shown in the inset of Fig. 1 suggests that other factors have contributed to the intensity.

X-ray diffraction pattern of the (1 − x)Pb(Mg1/2W1/2)O3 − xPb(Zr0.5Ti0.5)O3 ceramics. The inset reveals the (½ ½ ½) superlattice peak

Calculated (½ ½ ½) superlattice peak intensity based on the Pb[(Mg(1−x)Zrx)1/2(W(1−x)Tix)1/2]O3 model for the 1:1 cation order
Close examination of the pseudocubic peaks (200) and (222), as seen in Fig. 3, reveals a phase transition in the composition series. The ceramic with composition x = 0.93 takes a tetragonal symmetry which is isostructural to Pb(Zr0.5Ti0.5)O3, while those with compositions x = 0.60, 0.70, and 0.80 take an orthorhombic symmetry. The composition x = 0.90 appears to be the MPB composition. The observed symmetry change is consistent with the fact that Pb(Mg1/2W1/2)O3 crystallizes in an orthorhombic structure in the antiferroelectric state [8–12]. Furthermore, the structural order in the orthorhombic phase has been known to produce the (½ ½ ½) superlattice peak [8–12]. Therefore, the observed (½ ½ ½) superlattice peak in Fig. 1 has combined contributions from both the 1:1 cation order and the structural distortion order.

Close examination of (a) the pseudocubic (200) peak, and (b) the pseudocubic (222) peak of the (1 − x)Pb(Mg1/2W1/2)O3 − xPb(Zr0.5Ti0.5)O3 ceramics
In order to further clarify the origin of the (½ ½ ½) superlattice peak, one ceramic pellet of 0.4Pb(Mg1/2W1/2)O3 − 0.6Pb(Zr0.5Ti0.5)O3 was heated up to 500 °C and X-ray diffraction patterns were recorded at 150, 200, 300, and 500 °C. The temperature at dielectric peak, T m, of the composition x = 0.60 was determined to be 182 °C. Figure 4a shows the close view of the (½ ½ ½) superlattice peak while Fig. 4b shows the pseudocubic (200) peak at different temperatures. The crystal structure of the ceramic is expected to transform into the paraelectric phase with a cubic structure above T m. If this happens, the contribution from the structural order to the intensity of the (½ ½ ½) superlattice peak will be removed and the 1:1 cation order will be the sole source for the superlattice peak at temperatures above T m. As shown in Fig. 4a, the (½ ½ ½) superlattice peak indeed persists up to 500 °C without obvious weakening. However, the structure of the ceramic surprisingly remains a non-cubic structure even at 500 °C, as evidenced by the peak splitting around 44.5° shown in Fig. 4b. The result indicates that the high-temperature X-ray diffraction test is not capable of isolating the cation order contribution from the structural distortion to the superlattice peak intensity due to the non-cubic paraelectric phase.

Close examination of (a) the pseudocubic (½ ½ ½) superlattice peak, and (b) the pseudocubic (200) peak of the 0.40Pb(Mg1/2W1/2)O3 − 0.60Pb(Zr0.5Ti0.5)O3 ceramic at a series of temperature
The non-cubic paraelectric phase in 0.4Pb(Mg1/2W1/2)O3 − 0.6Pb(Zr0.5Ti0.5)O3 is highly unusual since the major component in the solid solution is Pb(Zr0.5Ti0.5)O3. It should be noted that non-cubic paraelectric phase was indeed reported previously in Pb(Mg1/2W1/2)O3 [8, 9] and Pb(Sc1/2Ta1/2)O3 [9]. The orthorhombic space group C2221 has been assigned to the paralelectric phase at temperatures above their Curie temperatures [8, 9]. It is interesting to notice that the structure remains non-cubic in the paraelectric phase even with a significant amount of Pb(Zr0.5Ti0.5)O3 added. Further detailed diffraction studies at high temperatures for the composition series are currently underway.
The dielectric constant of the composition series as a function of temperature during heating was measured at 1 kHz and is shown in Fig. 5. A broad dielectric peak is observed for most compositions. As x increases, the dielectric peak becomes sharper. The temperature at dielectric maxima, T m, increases with composition x, which is consistent with the high Curie temperature of Pb(Zr0.5Ti0.5)O3. The temperature T m, read from Fig. 5, is listed in Table 1. The dielectric maxima, ε m, increases with composition x and reaches the highest at x = 0.90. However, further increase in x to 0.93 leads to a significant drop. The high value of ε m in x = 0.90 can be attributed to the fact that it is the MPB composition.

Dielectric constant as a function of temperature measured at 1 kHz during heating in the (1 − x)Pb(Mg1/2W1/2)O3 − xPb(Zr0.5Ti0.5)O3 ceramics
The broad dielectric peaks shown in Fig. 5 resemble to those observed in relaxor ferroelectric ceramics [2–6]. Similar to the Curie–Weiss law for normal ferroelectrics, the dielectric constant for relaxor ferroelectrics can often be described by [20, 21]
where ε r is the dielectric constant above T m, T is the temperature, C′ is a constant, γ is the relaxation parameter. For typical relaxor ferroelectric like Pb(Mg1/3Nb2/3)O3, γ is about 2. When γ = 1, Eq. 1 becomes the Curie–Weiss law, describing a typical normal ferroelectric material such as BaTiO3 [20]. Fitting the dielectric constant data as shown in Fig. 5 determines the γ value in the (1 − x)Pb(Mg1/2W1/2)O3 − xPb(Zr0.5Ti0.5)O3 composition series. As seen from Table 1, it is interesting to notice that the highest relaxation parameter, γ, occurs in an intermediate composition x = 0.85. Such a non-monotonic change of γ with x seems to suggest that the intermediate compositions bear a stronger relaxor characteristic. This result indicates that a relaxor behavior can be realized by forming solid solutions between a normal ferroelectric and an antiferroelectric oxide. The observation strongly supports the previous model of relaxor ferroelectrics where the short range polar order is formed due to the competition between the long range antiferroelectric and the ferroelectric polar order [22]. The end member Pb(Mg1/2W1/2)O3 displays long range antiferroelectric order, while the end member Pb(Zr0.5Ti0.5)O3 shows long range ferroelectric order. In the composition of x = 0.85, the frustration of the long range polar order compromises at the nanometer scale short range polar order, leading to a strong relaxor behavior with a high relaxation parameter γ.
The broad dielectric peak and high γ values suggest a relaxor behavior in the solid solution (1 − x)Pb(Mg1/2W1/2)O3 − xPb(Zr0.5Ti0.5)O3. Such relaxor behavior is further confirmed by the frequency dispersion in the dielectric response in all the ceramics prepared. Figure 6 shows such frequency dispersion in compositions of x = 0.60 and 0.90. Again, the results support the hypothesis that a solid solution between an antiferroelectric and a ferroelectric may produce a relaxor ferroelectric. Similar frequency dispersion in dielectric response has been observed before in the (1 − x)Pb(Mg1/2W1/2)O3 − xPbTiO3 solid solution [23].

Dielectric constant and loss tangent as a function of temperature measured at 1, 10, and 100 kHz during heating in (a) the 0.40Pb(Mg1/2W1/2)O3 − 0.60Pb(Zr0.5Ti0.5)O3 ceramic, and (b) the 0.10Pb(Mg1/2W1/2)O3 − 0.90Pb(Zr0.5Ti0.5)O3 ceramic
Since all the compositions studied in this study display a relaxor ferroelectric behavior with a transition temperature that is much higher than room temperature, these ceramics can be considered as high temperature relaxor ferroelectrics, similar to the BiScO3–Pb(Mg1/3Nb2/3)O3–PbTiO3 ternary ceramics [24]. At room temperature, regular polarization versus field hysteresis loops may be observed due to the field-induced relaxor-to-ferroelectric phase transition [6]. This is indeed the case. A representative example for regular hysteresis loops is shown in Fig. 7a for composition x = 0.70. However, for the MPB composition x = 0.90, a pinched hysteresis loop is observed when the peak field is below 40 kV/cm (Fig. 7b). The hysteresis loop becomes normal at higher fields such as 60 kV/cm. The remanent polarization P r and coercive field E c were read directly from the hysteresis loops of all compositions measured at 40 kV/cm and are plotted in Fig. 8. The coercive field does not change significantly while the remanent polarization varies dramatically within the composition range. P r shows a minimum value in the composition x = 0.90 due to the distortion in the hysteresis loop.

Polarization versus electric field hysteresis loops measured at room temperature at 4 Hz in (a) the 0.30Pb(Mg1/2W1/2)O3 − 0.70Pb(Zr0.5Ti0.5)O3 ceramic, and (b) the 0.10Pb(Mg1/2W1/2)O3 − 0.90Pb(Zr0.5Ti0.5)O3 ceramic

Coercive field E c and remanent polarization P r read from hysteresis loops measured with a peak field of 40 kV/cm in the (1 − x)Pb(Mg1/2W1/2)O3 − xPb(Zr0.5Ti0.5)O3 ceramics
Piezoelectric coefficient d 33 was also assessed for compositions in the close vicinity of the MPB, specifically in compositions of x = 0.85, 0.90, and 0.93. The results are listed in Table 1. Interestingly, d 33 shows a monotonic increase with increasing Pb(Zr0.5Ti0.5)O3 content. In the ceramic of 0.07Pb(Mg1/2W1/2)O3 − 0.93Pb(Zr0.5Ti0.5)O3, d 33 is measured to be 219 pC/N.
Conclusions
The 1:1 B-site cation order, crystal structure, dielectric behavior, ferroelectric, and piezoelectric properties were studied in the (1 − x)Pb(Mg1/2W1/2)O3 − xPb(Zr0.5Ti0.5)O3 pseudo-binary solid solution system. It is suggested that both chemical and structural orders may have contributed to the presence of the (½ ½ ½) superlattice peak. An MPB between the orthorhombic and the tetragonal structures exists at x = 0.90. The dielectric behavior of the compositions studied (0.60 ≤ x ≤ 0.93) bears characteristics of relaxor ferroelectrics due to the competition between antiferroelectric and ferroelectric orders. In addition, a non-cubic perovskite structure was observed in the paraelectric phase above the dielectric maximum temperature T m. Regular hysteresis loops can still be measured at room temperature in these compositions due to a possible field-induced relaxor-to-ferroelectric phase transition. The best piezoelectric property at room temperature is measured in 0.07Pb(Mg1/2W1/2)O3 − 0.93Pb(Zr0.5Ti0.5)O3.
References
Mitchell RH (2002) Perovskite: modern and ancient. Almaz Press, Ontario
Smolenskii GA (1970) J Phys Soc Jpn 28(Suppl):26
Cross LE (1994) Ferroelectrics 151:305
Chen J, Chan HM, Harmer MP (1989) J Am Ceram Soc 72:593. doi:https://doi.org/10.1111/j.1151-2916.1989.tb06180.x
Davis PK, Akbas MA (2000) J Phys Chem Solids 61:159. doi:https://doi.org/10.1016/S0022-3697(99)00275-9
Zhao XH, Qu WG, He H, Vittayakorn N, Tan X (2006) J Am Ceram Soc 89:202. doi:https://doi.org/10.1111/j.1551-2916.2005.00675.x
Setter N, Cross LE (1980) J Appl Phys 51:4356. doi:https://doi.org/10.1063/1.328296
Zaslavskii AI, Bryzhina MF (1963) Sov Phys Crystallogr 7:577
Baba-Kishi KZ, Cressey G, Cernik RJ (1992) J Appl Cryst 25:477. doi:https://doi.org/10.1107/S0021889892001110
Baldinozzi G, Sciau P, Buffat PA (1993) Solid State Commun 86:541. doi:https://doi.org/10.1016/0038-1098(93)90135-A
Choo WK, Kim HJ, Yang JH, Lim H, Lee JY, Kwon JR et al (1993) Jpn J Appl Phys 32:4249. doi:https://doi.org/10.1143/JJAP.32.4249
Baldinozzi G, Sciau P, Pinot M, Grebille D (1995) Acta Crystallogr B 51:668. doi:https://doi.org/10.1107/S0108768194014047
Yasuda N, Fujimoto S, Yoshimura T (1986) J Phys C Solid State Phys 19:1055. doi:https://doi.org/10.1088/0022-3719/19/7/016
Baldinozzi G, Sciau P, Bulou A (1995) J Phys Condens Matter 7:8109. doi:https://doi.org/10.1088/0953-8984/7/42/008
Ardelean I, Barbur I, Timar V, Borodi Gh (2003) Mod Phys Lett B 17:1135. doi:https://doi.org/10.1142/S021798490300613X
Akbas MA, Davies PK (1997) J Am Ceram Soc 80:2933. doi:https://doi.org/10.1111/j.1151-2916.1997.tb03214.x
Juhas P, Davies PK (2004) J Am Ceram Soc 87:2086
Jaffe B, Cook WR, Jaffe H (1971) Piezoelectric ceramics. Academic Press, London
Shannon RD (1976) Acta Crystallogr A 32:751. doi:https://doi.org/10.1107/S0567739476001551
Uchino K, Nomura S (1982) Ferroelectr Lett 44:55. doi:https://doi.org/10.1080/07315178208201875
Vittayakorn N, Rujijanagul G, Tan X, Marquardt MA, Cann DP (2004) J Appl Phys 96:5103. doi:https://doi.org/10.1063/1.1796511
Chen IW (2000) J Phys Chem Solids 61:197. doi:https://doi.org/10.1016/S0022-3697(99)00282-6
Lu CH (1996) J Mater Sci 31:699. doi:https://doi.org/10.1007/BF00367888
Stringer CJ, Randall CA (2007) J Am Ceram Soc 90:1802. doi:https://doi.org/10.1111/j.1551-2916.2007.01640.x
Acknowledgement
This work was supported by the National Science Foundation through the CAREER grant DMR-0346819.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
White, D., Zhao, X., Besser, M.F. et al. Structure and properties of (1 − x)Pb(Mg1/2W1/2)O3 − xPb(Zr0.5Ti0.5)O3 solid solution ceramics. J Mater Sci 43, 5258–5264 (2008). https://doi.org/10.1007/s10853-008-2772-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-008-2772-1