
Abstract
The interaction between ciprofol and human serum albumin (HSA) was studied using spectroscopy-based approaches at different temperatures under simulated physiological conditions in vitro. Quenching of intrinsic Trp fluorescence of HSA with increasing ciprofol concentration is the actuating tool in the analysis. Experimental results proved that ciprofol quenched the intrinsic fluorescence of HSA through a static quenching mechanism. The thermodynamic parameters (ΔG = -2.35 × 104 J·mol−1, ΔS = -131 J·mol−1·K−1, and ΔH = -6.39 × 104 J·mol−1 at 310 K), binding sites (n = 0.83), and binding constant (KA = 9.12 × 103 M−1) indicated that hydrogen bond and van der Waals forces played a major role in the HSA-ciprofol association with weak binding force. Furthermore, the circular dichroism, synchronous, and three-dimensional fluorescence spectral results indicated adaptive structural changes of HSA in the presence of ciprofol. In addition, the effect of some common metal ions on the binding between ciprofol and HSA was examined, and Fe3+ and Hg2+ were proven to help prolong the storage time and improve the drug efficacy. The study provides accurate and full basic data for clarifying the binding mechanisms of ciprofol with HSA and helps understand its effect on protein function during the blood transportation process and activity in vivo.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ciprofol is a novel intravenous anesthetic compound closely resembling propofol in chemical structure. It exhibits rapid onset and recovery in preclinical studies[1]. Furthermore, ciprofol exhibited a lower free drug concentration in the aqueous phase than propofol under identical conditions and concentrations when analyzed using the “ultrafiltration method,” indicating a potential reduction or elimination of pain upon injection[2].
Serum albumin is the primary soluble protein in the circulatory system that plays various physiological roles, such as regulating blood osmotic pressure and pH, and acting as a carrier for transporting numerous endogenous and exogenous substances like drugs, amino acids, and fatty acids[3]. The interaction between drugs and serum albumin significantly influences the way drugs are distributed and their effectiveness[4]. Studying how pharmaceuticals interact with serum albumin is crucial for understanding drug nature, pharmacokinetics, and the correlation between drug structures and functions. Human serum albumin (HSA) consists of three structurally similar, mainly helical domains (I, II, and III), each with two subdomains (A and B)[5]. The main drug-binding areas on albumin are found in hydrophobic pockets in subdomains IIA and IIIA, which share similar chemical characteristics[6]. Consequently, the interaction between medicines and serum albumin consistently leads to interference with the binding of other pharmaceuticals due to overlapping binding sites or conformational changes.
Understanding the interactions between medications and serum albumin is crucial for comprehending pharmacokinetics and pharmacodynamics. A thorough study was conducted here to analyze the binding characteristics of ciprofol to HSA under standard physiological settings. We analyzed the binding information of HSA using various spectroscopic approaches to study quenching mechanisms, thermodynamic characteristics, binding modes, and conformation changes. The work offers precise and comprehensive foundational data to elucidate the binding methods of ciprofol with HSA. It aids in understanding the impact of drugs on protein function during the transportation processes in vivo.
Materials and methods
Materials
HSA (purity > 96%) was bought from Sigma Chemical Co. (St. Louis, USA) and was utilized without further purification. The HSA solutions were made in a pH 7.4 Tris − HCl buffer solution with a concentration of 0.2 mol/L, supplemented with NaCl at a concentration of 0.1 mol/L to maintain the solution's ionic strength. The HSA stock solution was diluted in Tris − HCl buffer right before use. Ciprofol with a purity of over 98% and all other reagents were bought from J&K Scientific Ltd. (Beijing, China). Ultrapure water was utilized in all the studies.
Fluorescence spectroscopy
The fluorescence spectra were measured using an F4500 spectrofluorophotometer (Hitachi, Tokyo, Japan) at various temperatures with slit widths set at 5 nm/5 nm. The excitation wavelength used was 295 nm for the excitation of Trp, and the emission spectra were measured within the range of 290 to 420 nm.
The synchronous fluorescence spectra were measured with a Δ-value of 15 or 60 nm between excitation and emission wavelengths. The emission and excitation bandwidths were both set at 5 nm. The spectra of HSA were recorded in the absence and presence of varying concentrations of ciprofol under similar conditions to steady-state fluorescence measurements.
Three-dimensional fluorescence spectra were conducted with the emission wavelength ranging from 200 to 500 nm. The excitation wavelength started at 200 nm with increments of 5 nm, ending at 400 nm. Other scanning parameters were identical to those used for the fluorescence quenching spectra.
UV–vis spectroscopy
UV − vis absorption spectroscopy was conducted using a UV-2450 UV − vis spectrophotometer from Shimadzu in Tokyo, Japan, which was fitted with a 1.0 cm quartz cell. The wavelength range was 290 to 420 nm.
Circular dichroism spectroscopy
Circular dichroism (CD) measurements were conducted with a JASCO J-815 automatic recording spectrophotometer from Tokyo, Japan, in a 1 cm quartz cell at room temperature. The concentrations of HSA and ciprofol were set at 2 × 10−7 and 1 × 10−6 M, respectively. Each spectrum was the mean of three consecutive scans. CD spectra were measured between 200 and 250 nm. The CDpro software suite (available at https://www.bmb.colostate.edu/cdpro/) was utilized to determine the secondary structural composition of HSA.
Results and discussion
Fluorescence measurements of macromolecules can provide insights into the interaction of small-molecule compounds with proteins, including details regarding the binding mechanism, mode, constant, sites, and intermolecular distances[7, 8]. Figure 1 displays the fluorescence emission spectra of HSA at pH 7.40 in the presence of ciprofol. When excited at 295 nm, HSA exhibited a prominent fluorescence emission peak at 336 nm. The fluorescence intensity of HSA diminished consistently as the concentration of ciprofol increased. Because the absorbance at both excitation and emission wavelengths of the interaction system was not strong enough (higher than 0.1), the decreased fluorescence intensity was not likely due to the inner filter effects but the interaction with ciprofol, indicating microenvironmental changes of Trp. The fluorescence quenching data were evaluated using the Stern–Volmer equation to confirm the quenching mechanism[9, 10]:
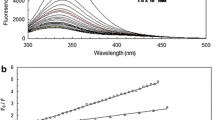
where F0 and F are the fluorescence intensity before and after the addition of the quencher, respectively, and Kq, KSV, τ0, and [Q] are the quenching rate constant, the Stern–Volmer quenching constant, the average lifetime of HSA (τ0 = 10−8 s) and the drug concentration, respectively. Table 1 displays the KSV and Kq values at various temperatures. The KSV values decreased with rising temperatures, indicating that the HSA-ciprofol quenching mechanism was static based on fluorescence quenching mechanisms (Fig. 2).

(A) Structure of HSA with Trp-214 shown. (B) Structure of the ciprofol molecule (C14H20O). Fluorescence spectra of HSA in the presence of different concentrations of ciprofol at 298 K. c(HSA) = 1 × 10−6 M and c(ciprofol, a-g) = 0, 1, 2, 5, 8, 12, and 15 × 10−6 M

(A) Stern–Volmer and (B) Hill plots for HSA-ciprofol at different temperatures. Conditions: pH 7.4; c(HSA) = 1 × 10−6 M
The binding constant (KA) and number of binding sites (n) for the ciprofol–HSA were calculated by using the following equation[11, 12]:
\(\lg \frac{F_0 - F}{F} = \lg K_A + n\lg [Q]\).
The binding constant increases as the temperature falls, but the value of (n) remains constant at different temperatures. The results suggest that a stable HSA-ciprofol complex at 310 K (physiological temperature) may be more stable than at a lower temperature. Table 1 displays the binding properties acquired at various temperatures. The n and KA values, derived from the linear regression equation of log (F0–F)/F versus log [Q], were determined to be 0.83 and 9.12 × 103 M−1, respectively. These findings suggest weak binding forces with fewer than one binding site at physiological temperature[8, 13, 14].
The determination of the acting force between ciprofol and HSA.
The following equations provided were used to assess the alterations in entropy, enthalpy, and free energy at different temperatures[15,16,17]:
where ΔH, ΔG, and ΔS denote enthalpy change, free enthalpy change, and entropy change, respectively. K represents the binding constant at the corresponding temperature. R represents the gas constant with a value of 8.314 J/mol/K, whereas T is the experimental temperature in Kelvin. A direct correlation was noted between the connection of lnK with 1/T, with a slope equivalent to the ΔH/R value (Fig. 3). When ΔH is negative and ΔS is positive, the major force is electrostatic interactions. When ΔH is negative and ΔS is negative, the main force is van der Waals or hydrogen bonding. When ΔH and ΔS are both positive, the main force is hydrophobic interactions. The table in Table 1 displays the values of ΔH, ΔG, and ΔS. The negative values of ΔH and ΔS indicate that hydrogen bonding and van der Waals forces are the major binding forces in the binding process.

van't Hoff plot of the HSA-ciprofol system
Energy transfer between ciprofol and HSA
Fluorescence resonance energy transfer (FRET) theory allows for the calculation of energy transfer parameters using the following equations. The efficiency of energy transfer (E) can be determined as[18]:
\(E = 1 - \frac{F}{F_0 } = \frac{R_0^6 }{{R_0^6 + r^6 }}\),
where E is the energy transfer efficiency between the donor and acceptor, and r is the binding distance between them. The equation for R0, the critical distance, where the transfer efficiency is 50%, is as follows[19]:
\(R_0^6 = 8.8{{ \times }}10^{ - 25} k^2 N^{ - 4} \Phi J\),
where k2 represents the orientation factor for the donor and acceptor of dipoles, N represents the refractive index of the medium, and Φ represents the fluorescence quantum yield of the donor. J represents the overlap integral of the fluorescence emission spectrum of the donor and the absorption spectrum of the acceptor and can be calculated using the following formula[20]:
\(J = \frac{{\int {F(\lambda )\varepsilon {{(}}\lambda {{)}}\lambda^4 {{\Delta }}\lambda } }}{{\int {F(\lambda ){{\Delta }}\lambda } }}\).
F(λ) represents the fluorescence intensity of HSA at wavelength λ, while ε(λ) stands for the extinction coefficient of the acceptor at λ. Figure S1 demonstrates a notable overlap between the emission spectrum of HSA and the absorption spectrum of ciprofol, indicating the potential for energy transfer between both molecules. For HSA, the values are k2 = 2/3, N = 1.336, and Φ = 0.118[21]. The equations provided yield the following results: J = 1.18 × 10–14 cm3 L mol−1, E = 0.098, R0 = 2.67 nm, and r = 2.98 nm. The r value falls within the range of 2–8 nm, with 0.5R0 < r < 1.5R0, suggesting an interaction between ciprofol and HSA.
Conformational studies by circular dichroism, 3D, and synchronous fluorescence spectroscopy
A circular dichroism (CD) analysis was conducted to investigate the protein structures. CD spectra of HSA and HSA–ciprofol were obtained using a CD spectrometer at room temperature within the wavelength range of 190 to 260 nm. The samples were examined in the far-ultraviolet range using a quartz cuvette with a route length of 1 cm. Figure 4 displays the second structure of the HSA curve and corresponding data. The CD spectra of HSA show two wide dip bands at 208 and 222 nm, indicating the transition of π − π* and n − π* of the α-helical structure of the HSA molecule. Alterations in the ellipticity at 208 and 222 nm are valuable indicators for observing changes in α-helical content[22]. The α-helix structure in HSA was 55%, closely matching Song et al.'s findings[23]. An increase in ciprofol concentration led to a decrease in the α-helix content of HSA compared to native HSA, indicating a change in the secondary structure of HSA due to the interaction with ciprofol. The β-turn content in HSA rose with the addition of ciprofol. The results show that ciprofol was strongly bound to HSA, which caused a change in its structure. The observed decrease in α-helical content and increase in β-content and random coiling indicate the disruption of the hydrogen bond network, which previously upheld the helical structure and overall skeleton. Due to the structural stability of β-sheets compared to α-helices, these changes in secondary structure may be linked to adaptive and irreversible modifications that counteract the disturbance generated by ciprofol[24].

(A) CD spectra of HSA (2 × 10−7 M) in the absence and presence of ciprofol (1 × 10−6 M). (B) The corresponding secondary structural fractions
We further investigated HSA conformational changes using 3D fluorescence spectroscopy. Comparative data can provide insights into how the protein's structure is affected due to microenvironmental changes. Alterations in the excitation or emission wavelength around the fluorescence peak, the introduction of new peaks, or the vanishing of existing peaks might provide valuable insights into the protein's structural changes. Excitation of HSA at 280 nm reveals the intrinsic fluorescence of Trp and Tyr residues, with minimal contribution from Phe residues. Figure 5 displays the 3D spectra and contour maps of HSA − ciprofol complexes, while Table 2 shows the spectral parameters obtained from these spectra. Peak I represents the spectrum characteristics of the fluorophore residues. Comparing it with the absorption spectrum of HSA, we may deduce that the peak at 280 nm is due to π − π* transitions of the aromatic amino acids. Peak II displays the protein's skeletal structural information. There was no Stokes shift but a significant drop in intensity for both peaks, suggesting changes in the surroundings of both fluorophores and the polypeptide backbone.

3D fluorescence spectra of pure HSA (A) and HSA-ciprofol (B). Conditions: pH 7.4; c(HSA) = 1 × 10−6 M; c(ciprofol): 1 × 10−5 M
Synchronous fluorescence screening is a crucial approach for gathering information about the chemical environment and its connection with chromophore molecules. Synchronous fluorescence spectroscopy was used here to investigate the impact of ciprofol binding on the conformation of HSA fluorophores and the molecular environment. The synchronous fluorescence spectra reveal precise details about the microenvironment of Trp or Tyr residues when measured at 60 or 15 nm[25]. Synchronous fluorescence spectra of HSA were observed both with and without ciprofol. Figure 6A shows only a 2 nm shift in the peak wavelength of Trp, suggesting slight alterations in the surrounding environment of the Trp residue. The Tyr synchronous fluorescence range remained constant, indicating no shift in polarity around the Tyr residue (Fig. 6B).

Synchronous fluorescence spectra of ciprofol with HSA with (A) Δλ = 15 nm or (B) Δλ = 60 nm at 310 K. Concentrations of the HSA and ciprofol are in line with Fig. 1
Effect of metal ions on HSA-ciprofol association
Plasma reportedly contains metal ions that can uphold the regular structures and physiological functions of proteins. Metal ions are necessary for humans and animals, playing a crucial structural function in many protein-based coordination connections[26]. Metal ions in plasma can influence the interaction of medicines with HSA. Cation concentrations were set at 1.0 × 10–5 M, and the anions were all chloride ions, which did not influence protein fluorescence. The binding constants of ciprofol with HSA at 310 K are included in Table 3. As shown in Table 3, The binding affinities between ciprofol and HSA were altered to different extents in the presence of metal ions, possibly due to conformational alterations around the binding sites. Reduced binding constants manifest decreased storage duration, which may increase harmful effects. Increased binding constants usually mean that the drug concentration in the circulation is buffered and its duration in the plasma is prolonged. Hence, the intended therapeutic outcome is attained. It can be inferred from the results that the elevated binding constants of HSA-ciprofol in the presence of Fe3+ and Hg2+ extend the drug's storage period in blood plasma and improve its maximal effectiveness.
Conclusions
In this research, various spectroscopic techniques have been used to explore the binding mechanisms of ciprofol to HSA. A static quenching process of the quenching mechanism of HSA induced by ciprofol has been revealed. The thermodynamic parameters manifested the critical role of hydrogen bond and van der Waals forces overwhelmed in keeping the stability of the ciprofol–HSA complex. The distance of ciprofol to HSA was close enough (r = 2.98 nm) to realize non-radiative energy transfer from Trp to ciprofol. As was illustrated by CD, 3D, and synchronous fluorescence results, the ciprofol binding induced microenvironmental changes of the protein and adaptive secondary structural changes featured by decreased α-helical content. Besides, Fe3+ and Hg2+ ions could enhance the ciprofol binding and prolong the blood storage time, which may improve the curative effect. This study is expected to facilitate further understanding of the interaction mechanism of HSA with ciprofol, which helps to provide theoretical foundations for ciprofol as an alternative to other phenol derivatives, such as propofol or fospropofol.
References
Hung, K.-C., Chen, J.-Y., Wu, S.-C., Huang, P.-Y., Wu, J.-Y., Liu, T.-H., Liu, C.-C., Chen, I.-W., Sun, C.-K.: A systematic review and meta-analysis comparing the efficacy and safety of ciprofol (HSK3486) versus propofol for anesthetic induction and non-ICU sedation. Front. Pharmacol. 14, 1225288 (2023)
Teng, Y., Ou, M., Wang, X., Zhang, W., Liu, X., Liang, Y., Li, K., Wang, Y., Ouyang, W., Weng, H.: Efficacy and safety of ciprofol for the sedation/anesthesia in patients undergoing colonoscopy: phase IIa and IIb multi-center clinical trials. Eur. J. Pharm. Sci. 164, 105904 (2021)
Zhao, X., Li, P., Song, S., Wang, H., Zhao, L., Zong, W., Zhang, H., Qu, G., Hu, L., Cai, Z.: Molecular structural heterogeneity of bisphenols governs their serum albumin binding. Sci. Total Environ. (2021). https://doi.org/10.1016/j.scitotenv.2021.146499
Sułkowska, A.: Interaction of drugs with bovine and human serum albumin. J. Mol. Struct. 614, 227–232 (2002)
Zhao, X., Sheng, F., Zheng, J., Liu, R.: Composition and stability of anthocyanins from purple solanum tuberosum and their protective influence on Cr(VI) targeted to bovine serum albumin. J. Agric. Food Chem. 59, 7902–7909 (2011)
Leboffe, L., di Masi, A., Polticelli, F., Trezza, V., Ascenzi, P.: Structural basis of drug recognition by human serum albumin. Current Med. Chem. (2019). https://doi.org/10.2174/0929867326666190320105316
Zhao, X., Hao, F., Lu, D., Liu, W., Zhou, Q., Jiang, G.: Influence of the surface functional group density on the carbon-nanotube-induced α-chymotrypsin structure and activity alterations. ACS Appl. Mater. Interf. 7, 18880–18890 (2015)
Mokaberi, P., Babayan-Mashhadi, F., Amiri Tehrani Zadeh, Z., Saberi, M.R., Chamani, J.: Analysis of the interaction behavior between Nano-Curcumin and two human serum proteins: combining spectroscopy and molecular stimulation to understand protein-protein interaction. J. Biomol. Struct. Dynam. 39, 3358–3377 (2021)
Zhao, X.C., Liu, R.T., Chi, Z.X., Teng, Y., Qin, P.F.: New insights into the behavior of bovine serum albumin adsorbed onto carbon nanotubes: comprehensive spectroscopic studies. J. Phys. Chem. B 114, 5625–5631 (2010)
Zhu, Y., Liu, G., Liu, A., Yu, L.: biophysical study on the interaction of dexmedetomidine and serum protein. Lat. Am. J. Pharm. 33, 1669–1677 (2014)
Zhao, X., Lu, D., Hao, F., Liu, R.: Exploring the diameter and surface dependent conformational changes in carbon nanotube-protein corona and the related cytotoxicity. J. Hazard. Mater. 292, 98–107 (2015)
Chanphai, P., Tajmir-Riahi, H.: Tea polyphenols bind serum albumins: a potential application for polyphenol delivery. Food Hydrocoll. 89, 461–467 (2019)
Rahnama, E., Mahmoodian-Moghaddam, M., Khorsand-Ahmadi, S., Saberi, M.R., Chamani, J.: Binding site identification of metformin to human serum albumin and glycated human serum albumin by spectroscopic and molecular modeling techniques: a comparison study. J. Biomol. Struct. Dyn. 33, 513–533 (2015)
Danesh, N., Navaee Sedighi, Z., Beigoli, S., Sharifi-Rad, A., Saberi, M.R., Chamani, J.: Determining the binding site and binding affinity of estradiol to human serum albumin and holo-transferrin: fluorescence spectroscopic, isothermal titration calorimetry and molecular modeling approaches. J. Biomol. Struct. Dyn. 36, 1747–1763 (2018)
Beigoli, S., Sharifi Rad, A., Askari, A., Assaran Darban, R., Chamani, J.: Isothermal titration calorimetry and stopped flow circular dichroism investigations of the interaction between lomefloxacin and human serum albumin in the presence of amino acids. J. Biomol. Struct. Dyn. 37, 2265–2282 (2019)
Kamshad, M., Jahanshah Talab, M., Beigoli, S., Sharifirad, A., Chamani, J.: Use of spectroscopic and zeta potential techniques to study the interaction between lysozyme and curcumin in the presence of silver nanoparticles at different sizes. J. Biomol. Struct. Dyn. 37, 2030–2040 (2019)
Sadeghzadeh, F., Entezari, A.A., Behzadian, K., Habibi, K., Amiri-Tehranizadeh, Z., Asoodeh, A., Saberi, M.R., Chamani, J.: Characterizing the binding of angiotensin converting enzyme I inhibitory peptide to human hemoglobin: Influence of electromagnetic fields. Protein Pept. Lett. 27, 1007–1021 (2020)
Chamani, J.: Energetic domains analysis of bovine α-lactalbumin upon interaction with copper and dodecyl trimethylammonium bromide. J. Mol. Struct. 979, 227–234 (2010)
Cheng, Z.: Comparative studies on the interactions of honokiol and magnolol with human serum albumin. J. Pharm. Biomed. Anal. 66, 240–251 (2012)
Meti, M.D., Byadagi, K.S., Nandibewoor, S.T., Chimatadar, S.A.: Multi-spectral characterization & effect of metal ions on the binding of bovine serum albumin upon interaction with a lincosamide antibiotic drug, clindamycin phosphate. J. Photochem. Photobiol., B 138, 324–330 (2014)
Lu, D.W., Zhao, X.C., Zhao, Y.C., Zhang, B.C., Zhang, B., Geng, M.Y., Liu, R.T.: Binding of Sudan II and Sudan IV to bovine serum albumin: comparison studies. Food Chem. Toxicol. 49, 3158–3164 (2011)
Zhao, X., Lu, D., Liu, Q.S., Li, Y., Feng, R., Hao, F., Qu, G., Zhou, Q., Jiang, G.: Hematological effects of gold nanorods on erythrocytes: hemolysis and hemoglobin conformational and functional changes. Adv. Sci. 4, 1700296 (2017)
Song, S., Li, Y., Liu, Q.S., Wang, H., Li, P., Shi, J., Hu, L., Zhang, H., Liu, Y., Li, K.: Interaction of mercury ion (Hg2+) with blood and cytotoxicity attenuation by serum albumin binding. J. Hazard. Mater. (2021). https://doi.org/10.1016/j.jhazmat.2021.125158
Leuenberger, P., Ganscha, S., Kahraman, A., Cappelletti, V., Boersema, P.J., von Mering, C., Claassen, M., Picotti, P.: Cell-wide analysis of protein thermal unfolding reveals determinants of thermostability. Science 355, eaai825 (2017)
Zhao, Z., Li, H., Yao, J., Lan, J., Bao, Y., Zhao, L., Zong, W., Zhang, Q., Hollert, H., Zhao, X.: Binding of tetrabromobisphenol A and S to human serum albumin is weakened by coexisting nanoplastics and environmental kosmotropes. Environ. Sci. Technol. 57, 4464–4470 (2023)
Zhao, Z., Yao, J., Li, H., Lan, J., Hollert, H., Zhao, X.: Interaction of polystyrene nanoplastics and hemoglobin is determined by both particle curvature and available surface area. Sci. Total Environ. 899, 165617 (2023)
Author information
Authors and Affiliations
Contributions
Q.P. and C.Y. wrote the main manuscript text and Y.Z. prepared all the figures. S.S. guided the work. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Pan, Q., Yao, C., Zhu, Y. et al. Probing the interaction of ciprofol and human serum albumin using multiple spectroscopies. J Incl Phenom Macrocycl Chem 104, 407–414 (2024). https://doi.org/10.1007/s10847-024-01241-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10847-024-01241-5