
Abstract
Chelation and solution thermodynamic stability of a tripodal hydroxypyranone-based chelator (tris[(5-hydroxy-4-oxo-pyran-2-yl)methyl]benzene-1,3,5-tricarboxylate), TBHPY, towards biologically relevant divalent metal ions:Cu(II), Fe(II), Ni(II), Co(II) and Zn(II) were studied by potentiometric and spectroscopic methods in 9:1 (H2O:DMSO) medium. The metal ions formed ML, MLH2, MLH, MLH-2, and MLH-1 type complexes with high formation constants. The ligand was explored for its application as a potential fluorimetric sensor and examined in the presence of various cations. Nearly twofold quenching was observed upon addition of Gd(III) to TBHPY. The experimental, spectroscopic and thermodynamic stability results were validated with the theoretical quantum mechanical calculations using density functional theory (DFT). The geometrical structures, electronic properties,and bonding behavior of the complexes are described in detail.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Heterocycles play a crucial role in medicinal and organic chemistry by bridging biochemistry and life science [1]. Among the heterocycles, hydroxypyridinones and hydroxypyranones play significant roles in drug design. The derivatives of such compounds exhibit a range of biological actions, including anti-diabetic, anticonvulsant, antioxidant, antiviral, anticancer, antifungal, anti-inflammatory, and antibacterial activities [2]. Hydroxypyridinones and hydroxypyranones are potential bidentate chelating agents with bonding through ketonic and hydroxylic oxygen atoms at appropriate positions on heterocyclic rings, and their corresponding anions form relatively stable complexes with various metal ions [3]. Kojic acid, (5-hydroxy-2-hydroxymethylpyran-4-one), (Fig. 1a) which belongs to the family of hydroxypyranones is widespread for its applications in various places like food, pharmaceuticals, cosmetics, and chemical industry and agriculture. While there is remarkable applications of kojic acid in various fields, its advantages in the medical industry and cosmetics are more noteworthy [4]. The chelating behavior of kojic acid and its derivatives is significant for their antifungal, antineoplastic, and antimicrobial activity [5, 6]. Siderophores, a group of natural chelators that bind iron, form stable complexes with high binding constants and transport in cell membranes. Among the known natural siderophores, enterobactin has the highest formation constant with log β = 1049 [7]. It is a hexadentate chelator that forms an octahedral iron complex by binding through three bidentate catecholate groups linked through amide spacers to a central cyclic L-serine unit. Many siderophore-mimics have been synthesized [8,9,10,11,12,13,14,15] by taking advantage of the high binding constant and its unique structure.The efficacy and chelation therapy properties of the described hydroxypyranone or hydroxypyridinones can be multiplied by garlanding many such elements into a single unit. Several such siderophore-mimics have been synthesized and reported [16,17,18,19,20,21,22]. A wide variety of diseases were concomitant with an improper balance of metal ion levels in the body, including diabetes, neurodegenerative diseases, and cancer [23,24,25]. Iron, copper, and zinc are significant for living beings and are linked with several biomolecules associated with crucial physiological activities [26,27,28]. Cobalt is one of the essential metals comprising the active center of the coenzyme cobalamin, of which vitamin B12 is the most trivial [29]. A case was reported by Andersen in Canada about acute intoxication from cobalt salts [30]. Cobalt is a leading source of contact dermatitis after nickel and chromium [31]. Distinct cobalt species are listed as possible carcinogens by IARC [32]. Nickel is also significant regarding health and some related diseases [33, 34]. Some evidence has recommended that Ni ion imparts a significant role in hematopoiesis along with vitamin B12 [35, 36]. Cardiovascular system poisoning, lung fibrosis, cancer, and skin allergies are developed due to long-term exposure to nickel [37].

Depiction of structures of a Kojic acid and b TBHPY (tris[(5-hydroxy-4-oxo-pyran-2-yl)methyl] benzene-1,3,5-tricarboxylate)
In continuation to our work on the tripodal chelator tris[(5-hydroxy-4-oxo-pyran-2-yl)methyl] benzene-1,3,5-tricarboxylate (TBHPY) (Fig. 1b) [16], in this paper, studies on solution thermodynamic stability constants of the TBHPY with Fe(II), Co(II), Ni(II), Cu(II), and Zn(II) by spectrophotometric and potentiometric methods are described. DFT calculations were also performed to establish the geometrical structures, electronic and spectroscopic properties, and bonding nature of the complexes. The applicability of the ligand use of fluorescence probe for metal ions was also explored.
Results and discussion
Solution thermodynamics and complex formation equilibria
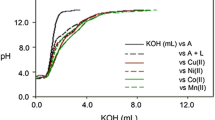
The complex formation activities of TBHPY with the divalent metal ions Fe(II), Co(II), Ni(II), Cu(II) and Zn(II) were probed by spectrophotometric and potentiometric methods. Potentiometric titrations were performed in a 1:1 (M:L) molar ratio in highly aqueous media (9:1:: H2O:DMSO) due to solubility issues, at 298 K and an ionic strength of μ = 0.1 M KCl. The metal–ligand titration curves showed deviations at the lower pH region (< 4) from the ligand curve, suggesting complex formation with the metal ion (Fig. 2). The titration data were fitted in a global fitting program Hyperquad [38] for refining of stability constants. Several models were considered in the fitting program in which the best-fit model was obtained by considering the formation of five complex species: ML, MLH2, MLH, MLH-2 and MLH-1. At lower pH (4–5), protonated MLH2 species formed predominantly for all the divalent ions; with the increase in pH, MLH and ML species started forming, and, with a further increase in the pH two hydrolysis species MLH-1 and MLH-2 were resulted. In the case of iron, FeLH species were formed with 85% at neutral pH followed by negligible formation of ML, and, at higher pH, hydrolysis occurred. For cobalt, CoL (40%) and CoLH (2–3%) formation occurred after pH 5, and ultimately, CoLH-1 existed as the predominant species at pH ~ 7–8. The second hydrolysis species CoLH-2 was formed with the further rise in pH. NiL species was predominant in nickel, with 50% formation at pH 6.8, whereas NiLH species remained nearly 25% at pH 6.4. Afterward, hydrolysis species formed with NiLH-1 species at pH 7.2 with nearly 25% formation. CuL predominantly formed with 70% at nearly pH 7.

Potentiometric titration curve of ligand and its Fe, Co, Ni, Cu, and Zn complexes in 1:1 (M:L) ratio in 9:1 (H2O:DMSO) medium at 298 K and µ = 0.1 M KCl
At pH 6, CuLH species were observed with nearly 20% formation. In the case of zinc, ZnLH was formed at pH 7.3 with nearly 40% existence. Afterward, ZnL was formed predominantly with 60% at pH 7.7. Further increments in pH resulted in hydrolysis species ZnLH-1, with nearly 20% at pH 8, and ZnLH-2 was predominantly present at higher pH levels.The percentages of individual species formed as a function of pH were obtained by employing the refined formation constants (log K) in the program Hyss [39] for the metal-complex systems are shown in Fig. 3. The log Ks of ML complexes of Fe(II), Co(II), Ni(II), Cu(II) and Zn(II) followed the expected Irving-Williams order. The ML complexes exist in the pH range ~ 5 to ~ 9 with maximum % of ML complex formation appeared at about pH ~ 7 for iron, cobalt, nickel and copper complexes, whereas for zinc complex the maximum % was observed at pH 7.7. The slight variation in pH for the formation of complexes may be explained on the basis of the unequal difference in the stability constants of protonated and hydroxy complexes with ML, that ascended due to changes in the electronic behaviour.

Species distribution curves of M-L complexes as a function of pH with [M] = 10–3 M where M is (a) Fe(II) (b) Co(II) (c) Ni (II) (d) Cu (II) and (e) Zn (II), at T = 298 K and µ = 0.1 M KCl
The plausible solution structure of ML has been predicted by the molecular modeling studies (Sect. "Molecular modeling and computational methods"). Based on that, the structures of complex species ML, MLH2, MLH, MLH−2 and MLH−2 with their possible coordination modes are presented in Fig. 4. The MLH−1 and MLH−2 are equivalent to ML(OH) and ML(OH)2 respectively, that were obtained from the hydrolysis of the complexes of the coordinated water molecules at higher pH. The electrical charges in all the species are ignored here for the sake of simplicity. For further validation of the formation of different species and the stability constants in the solution obtained from potentiometric studies, spectrophotometric titrations with changes in pH were employed (Fig. 5). TBHPY shows an absorption peak at 272 nm in the UV–visible region attributed to π → π* transition [16]. There is a considerable change in the spectra upon coordination with the metal ions. The prominent ligand π → π* band remains almost intact in the lower pH ~ 3.5 in all the complexes. The λmax values were found at 270 nm (ε = 2.72 × 104 Lmol−1 cm−1), 271 nm (ε = 2.51 × 104 Lmol−1 cm−1), 273 nm (ε = 2.01 × 104 Lmol−1 cm−1), 271 nm (ε = 2.78 × 104 Lmol−1 cm−1) and 273 nm (ε = 2.55 × 104 Lmol−1 cm−1) for Fe (II), Ni (II), Co (II), Zn (II) and Cu (II) systems, respectively. Upon an increase in pH, these absorption bands shifted to a higher wavelength, which was prominently noticeable at and after pH 7.The intensity of the electronic absorption spectra at lower pH decreased upon successive addition of KOH, while a regular increase in the intensity for the absorption band occurred at higher pH.

Probable molecular structures of the complex species formed by interaction of TBHPY with divalent metal ions (M = Fe(II), Co(II), Ni (II), Cu (II) and Zn (II)) in solution

Spectrophotometric titration curves of TBHPY with different metal ions within the pH range of 2–10 (a) TBHPY–Fe(II) (b) TBHPY–Co(II) (c) TBHPY–Ni(II) (d) TBHPY–Cu(II) and (e) TBHPY–Zn(II)
The log β values for all the divalent metal ion complexes of the ligand TBHPY were evaluated by potentiometric (A) and spectrophotometric (B) methods (as listed in Table 1) and compared with other hydroxypyranone based scaffolds listed in Table 2.
The comparison table depicted that log β values evaluated for the ligand are comparable with the reported analogs of hydroxypyranones. The log β values of MLH2, MLH, ML, and MLH-1 species of TBHPY were comparable to those of already reported tripodal ligand (2) (Table 2). The tripodal ligand (2) has complexes with a higher stability constant than TBHPY. This may be attributed to the flexible nature of the central TREN unit. The ligand (1) has a lower log β value for ZnL than others because of two hydroxypyranone moieties, whereas TBHPY and (2) comprise three hydroxypyranone units. In the case of other reported ligands, ligand (1) has log β = 25.38 for CuLH2 species and log β = 18.17 for ZnLH species, whereas TBHPY has log β = 31.69 for CuLH2 species and log β = 24.60 for ZnLH species. The trend of stability constants of the reported transition metal complexes follows the “Irving–Williams stability order”, Fe(II) < Co(II) < Ni(II) < Cu(II) > Zn(II) [40].
1: 6,6′-(2-(diethylamino)ethylazanediyl)bis(methylene)bis(5-hydroxy-2-hydroxymethyl-4H-pyran-4-one); 2: 6,6′,6″-(((nitrilotris(ethane-2,1-diyl))tris(azanediyl))tris(methylene))tris(3-hydroxy-4H-pyran-4-one); 3: 2,2’-[ethane-1,2-diylbis(iminomethanediyl)]bis(5-hydroxy-4H-pyran-4-one); 4: 2,2’-[propane-1,3-diylbis(iminomethanediyl)]bis(5-hydroxy-4H-pyran-4-one); 5: 2,2’-[butane-1,4-diylbis(iminomethanediyl)]bis(5-hydroxy-4H-pyran-4-one).
Application of TBHPY as potential fluorimetric sensor
The ligand TBHPY is expected to exhibit fluorescence under suitable conditions. When excited at 270 nm, the ligand demonstrated fluorescence emission at ~ 350 nm in DMSO solution at room temperature. There is also a possibility that some cations may alter the fluorescence of the ligand, thereby affecting its applicability as a fluorescence probe. Cations such as Na(I), K(I), Co(II), Fe(III), Cd(II), Cr(III), Cu(II), Fe(II), Al(III), Mn(II), Mg(II), Pb(II), Hg(II), Ni(II), Zn(II), Eu(III), Gd(III), Tb(III) were chosen for study. The absorption spectra of the 1:1 metal–ligand for the above metal ions were taken for the photophysical studies in 0.5 × 10–4 DMSO at room temperature. Figure 6a depicts the changes in the absorption spectra by the addition of metal ions. An absorption band at ~ 272 nm was observed for the ligand and the metal-TBHPY mixture. A shoulder was observed at 325 nm, 336 nm, 347 nm or 325 nm for Fe(III), Cu(II), Ni(II), and Cd(II) respectively. The dominance of π → π* transitions can be depicted from the electronic spectra. Systematic fluorescence studies were done to find the selectivity of TBHPY with different metal ions. No significant changes in the intensity of fluorescence were reported in the presence of all metal cations except Gd(III) (Fig. 6b). Gd(III) quenched nearly twofold time upon addition to TBHPY, indicating the use of later as a probe for trivalent gadolinium. This quenching in fluorescence may be attributed to chelation-enhanced quenching (CHEQ), which can result after the formation of a rigid complex between TBHPY-Gd(III).

Absorbance (a) and Fluorescence spectra (b) of TBHPY at 0.5 × 10–4 M in the presence of various metal ions at 0.5 × 10–4 M in DMSO
Molecular modeling and computational methods
To gain a deep understanding of the bonding, molecular structures, spectral and electronic properties of the metal complexes, molecular modeling studies were performed using DFT in gas phase. The structures were initially optimized by molecular mechanics, semi-empirical, and then finally utilizing DFT method [44,45,46] with GGA/PBE-D3 functional and TZP basis sets.
Molecular structures
The DFT-optimized structure of TBHPY presented an extended geometry where two hydroxypyranone units were undergoing face-to-face π-stacking [16]. The divalent metal ions can form complexes by coordinating the ligand with the metal ions as hexadentate plausible coordination mode employing three bidentate hydroxypyranone units. To explain the structures of complexes, a possible hexadentate mode of the ligand was proposed, but binding with six oxygens of hydroxypyranone to the same metal ion was found to be energetically unfavorable. Modeling the structures suggests a square-pyramidal geometry for all metal ML complexes. It is known that metal complexes may display different structural geometry in the solid state or solution. In the presence of water as a solvent, the transition metal ions assume M(H2O)6n+ geometry. In the current case, the final proposed structure of ML was assumed to be ML(H2O), in which one of the O of ligand L is not coordinated.
Based on the above, the molecular formula of the complexes can be designated as [Fe(TBHPY)(H2O)], [Co(TBHPY)(H2O)], [Ni(TBHPY)(H2O)], [Cu(TBHPY)(H2O)] and [Zn(TBHPY)(H2O)] with a negative charge. The charge of the complexes are not shown here for the sake of simplicity.The structures were finally considered for DFT calculations. The structures of all ML complexes in gas phase optimized by GGA-PBE-D3//TZP method are shown in Fig. 7. All the coordination compounds display distorted octahedral geometries. The geometrical parameters are also presented in Table 3. The absence of any imaginary frequency in the optimized structures confirmed that they are at true minima.

DFT optimized structures of metal complexes of TBHPY a Fe–TBHPY b Co–TBHPY c Ni–TBHPY d Cu–TBHPY and e Zn–TBHPY obtained by PBE-D3/TZP method, f numbering scheme for Table 3
Frontier molecular orbital analysis
The electronic properties and the stability of a molecule can be derived from the Frontier Molecular Orbital (FMO) analysis, particularly from the HOMO and the LUMO. These molecular orbital diagrams of the frontier orbitals of the complexes are depicted in Fig. 8.

HOMO and LUMO frontier molecular orbitals of TBHPY metal complexes (a) Fe(TBHPY) (b) Co(TBHPY) (c) Ni(TBHPY) (d) Cu(TBHPY) (e) Zn(TBHPY)
In the complex [Fe(TBHPY)(H2O)], the HOMO (113 A) consists of a singly occupied MO (SOMO) having energy -3.037 eV, and the LUMO (114 A) with energy − 2.947 eV, leading to an energy gap of 0.090 eV. The HOMO is predominantly contributed from iron d-orbitals and comprises 70.36% of dxz, and 6.99% of dyz, with minor contributions from d-orbitals of Fe. In LUMO 114 A, a significant contribution of 22.65%, 16.27%, and 10.53% was observed from pz orbitals of C atoms of the benzene ring. In the complex [Co(TBHPY)(H2O)], the HOMO is 114 A having an energy of − 3.744 eV, and the LUMO is MO 115 A with energy − 3.148 eV, thereby resulting in 0.596 eV energy gap. The HOMO 114 A consists of a predominant contribution from d orbitals of Co with 38.17% of dyz, 33.94% of dxz, and 12.50% of dx2-y2 orbitals. 115 A, the LUMO of this cobalt complex imparts a contribution of 23.25%, 16.22%, 11.42%, and 10.25%, majorly from the pz orbitals of carbon atoms of the benzene ring. The [Ni(TBHPY)(H2O)] has MO 115A as HOMO with energy − 4.390 eV and 116 A as the LUMO with energy − 3.099 eV. The MO 115 A comprises a significant contribution from d orbitals of Ni with 31.03% from dx2-y2, 10.73% from dyz, and 14.70% from dxy orbitals. The LUMO 116 A has a predominant significant contribution from the pz orbitals of the C atom from the central unit as compared to its HOMO. In the case of [Cu(TBHPY)(H2O)], the energy of HOMO (MO 117 A) and LUMO (MO 118 A) is − 4.630 eV and − 3.068 eV, respectively. The HOMO is comprised of a contribution of 9.12% from Cu while 9.11% and 6.76% contribution are the results due to the py and pz orbital of the oxygen atom of hydroxypyranone moiety, respectively along with a contribution of 6.60% from the py orbital of C atom.
118 A orbital (LUMO) is contributed as a result of the contribution from the pz orbitals (20.48%, 19.72%, 9.83% and 9.41%) of C atoms from benzene ring of central moiety. 117 A and 118 A contributed as HOMO and LUMO, respectively, in the complex [Zn(TBHPY)(H2O)]. An energy gap of 1.851 eV is observed from the HOMO (− 4.832 eV) and LUMO energy (− 2.981). The HOMO has attained a major contribution from py orbitals of oxygen (11.26%) and carbon atoms (11.22% and 6.01%) of hydroxypyranone unit.The residual contribution is from the px, py, and pz orbitals of oxygen and carbon atoms, along with a minor contribution (1.24%) from the dxy orbital of Zn. The MO 118 A (LUMO) contributes predominantly from orbitals of carbon atoms (43.27% pz,, 14.60% pz, and 11.28% pz) of the benzene ring.
Electronic absorption spectra
The electronic absorption spectra in terms of intensity and the electronic transition properties of a molecule can be attained by a time-dependent density functional theory (TDDFT). The absorption spectra were calculated theoretically by TDDFT simulations using GGA-PBE as XC functional and TZ2P basis sets.Table 4 summarizes some relevant data related to the construction of molecular orbitals with percentage contribution from atomic orbitals. In the case of [Fe(TBHPY)(H2O)] absorption spectra (fcal = 0.0237), the band is displayed due to a transition from 113A_β → 118A_β with 65.2% contribution.
For [Co(TBHPY)(H2O)], the band (fcal = 0.0016) was observed corresponding predominantly contribution by transition from 114A_β → 118A_β (95.8%). In the case of [Ni(TBHPY)(H2O)], the absorption spectra (fcal = 0.0014) revealed that the major contribution is due to the transition from 115 A_β → 119A_β (73.3%) along with some minor contribution from 115A_β → 118A_β (21.7%). For the [Cu(TBHPY)(H2O)] complex, the absorption peak was observed corresponding to the transition from 112A_β → 117A_β (47.7%) and 113A_β → 117A_β (41.8%). The absorption spectra of [Zn(TBHPY)(H2O)] shows a band at 447.3 nm allotted to 117A → 120A transition with contribution of 78.0%.
Global chemical reactivity descriptors
The chemical reactivity and stability are usually described using global reactivity descriptorslike global hardness (η),electron affinity (A), electronegativity (χ), global softness (S), chemical potential (μ), ionization potential (I), and electrophilicity (ω) from the Kohn–Sham DFT [47,48,49]. All these parameters can be derived using Koopmans’ theorem and frontier molecular orbital energy [50]. The computed results acquired for these complexes were correlated with all the parameters mentioned earlier (Table 5). The negative chemical potentials (μ) values (− 2.99, − 3.44, − 3.74, − 3.84, and − 3.90, respectively) indicate that the Fe(II), Co(II), Ni(II), Cu(II), and Zn(II) complexes are quite stable enough and do not decompose into elements. The values of chemical hardness (0.045, 0.298, 0.645, 0.781, and 0.925) are well supported by the HOMO–
LUMO gap 0.090, 0.596, 1.291, 1.562, and 1.851 eV for Fe(II), Co(II), Ni(II), Cu(II) and Zn(II), respectively,suggesting that they are less polarizable because of the electron cloud resistance of a chemical system to deform under small perturbations.The values of global softness (22.22, 3.355, 1.550, 1.280, and 1.081 eV) have revealed that the Fe(II) complex is softer than other complexes. The other parameters are also presented in Table 5. The results from the energy gaps of the metal complexes indicate that the kinetic stability of the complexes follows the order: Fe(TBHPY) < Co(TBHPY) > Ni(TBHPY) < Cu(TBHPY) < Zn(TBHPY).
Experimental
Materials and instrumentation
The Sigma Aldrich and CDH chemicals were used as such without purifying them. UV–visible spectrophotometer (Evolution 201 Thermoscientific) was used to carry out spectrophotometric studies. The fluorescence studies were investigated using an Agilent Technologies Cary Eclipse fluorescence spectrophotometer. The potentiometric titrations were done in a highly aqueous medium, i.e., H2O:DMSO (9:1, v/v). pH measurements were performed using a Thermoscientific Orion star pH meter. The LINUX operating system on an Intel Core i7 CPU performed DFT calculations.
Potentiometric measurements
The nature of the coordination behavior of the ligand with its metal complexes was investigated potentiometrically using a double wall titration vessel at 298 K in highly aqueous media (H2O: DMSO:: 9:1). To perform the pH titrations, the pH meter having Ross Ultra pH electrode was used.The classical method was used for the calibration of the electrode to get correct pH values. HCl and KOH solutions (0.1 M) were standardized using KHP (0.1 M) to obtain the exact concentration.The potentiometric measurements were done with the 10–3 M solution of the ligand, and KCl was added to maintain the ionic strength μ = 0.1. The acidification of 10–3 M ligand solution was done upto pH 2 using 0.1 M HCl to ascertain the presence of any kind of species at a lower pH. The solution was titrated consistently against the standard KOH solution (0.1 M) to investigate the log K values within the 2–11 pH range. The ligand complexation behavior was inspected with Cu(II), Fe(II), Ni(II), Co(II), and Zn(II) upon titration of 1:1(M:L) solution against KOH (0.1 M). 10–3 M concentration solutions were prepared using sulfate salts of the metals as mentioned above. The final concentration was 10–3 M for ligand solution and metal ions solution used. Hyperquad 2006 [38] refinement program was used to explore the formation of different species and the determination of equilibrium constants. Hyss 2009 was used to evaluate the species distribution curve [39].
Spectrophotometric measurements
Spectrophotometric titrations were done in a similar manner as potentiometric titrations. The absorption spectrum was recorded by taking the solution in the cuvette every time KOH was added. The aliquot solution was then again put into the titration flask with precision. The global fitting program Hypspec was used to analyze the binding constants from titration data [51].
Photophysical studies
The fluorescence technique and UV–visible spectroscopic method were used to investigate the interaction between the ligand and various cations at room temperature in the DMSO medium. Stock solutions (10–3 M) of the ligands and metal ions were prepared using DMSO. Solutions of concentration 0.5 × 10–4 M were prepared using these stock solutions to analyze the absorption and emission properties.
Molecular modeling
All the calculations were carried out on an Intel Core i7 machine using Amsterdam Modeling Suite (AMS) software version 2022.103 [52]. All the geometries were optimized by DFT after obtaining the least strained geometry from molecular mechanics calculations [44,45,46]. These molecular geometries were optimized using GGA-PBE-D3 basis set in the gas phase, with dispersion correction. The vibrational frequencies were also calculated after geometry optimization of the molecules to further confirm that the resulting structures obtained relate to the true minima.
Conclusions
The reported TBHPY hexadentate ligand comprised of three hydroxypyranone units as binding moieties symmetrically attached to a central unit of benzene was studied for complexation with divalent metal ions (Cu, Co, Fe, Zn, and Ni). The complexation behavior of the ligand with these metal ions in solution depicted the formation of ML, MLH, and MLH2 species, along with two hydrolysis species for all the metal ions. The log β values of ML complexes followed the trend: Zn < Cu > Ni > Co > Fe with the highest value of log β = 19.28 for copper. These complexes can be greatly useful in chelation therapy and the treatment of metal overload diseases with divalent metal ions due to the high stability constants of their complexes compared to other reported analogs. In addition, the applicability of the TBHPY as probe for Gd(III) was confirmed from the fluorescence emission of the ligand in presence of a variety of cations viz., Hg(II), Na(I), Cu(II), Mn(II), Co(II), Cr(III), Cd(II), Fe(II), Al(III), Fe(III), Mg(II), Pb(II), Zn(II), K(I), Ni(II), Eu(III), Gd(III), Tb(III) due to its 2.5 fold quenching in DMSO solution. The DFT studies revealed the structure to be distorted octahedral with formulation [M(TBHPY)(H2O)]. The electronic spectral studies, either experimental or theoretical, depicted that transitions are predominantly π → π*. The high formation constants of TBHPY complexes signify that the ligand can be explored further for its employability as a promising chelator in chelation therapy or metal sequestration.
References
He, M., Fan, M., Peng, Z., Wang, G.: An overview of hydroxypyranone and hydroxypyridinone as privileged scaffolds for novel drug discovery. Eur. J. Med. Chem. 221, 113546 (2021). https://doi.org/10.1016/j.ejmech.2021.113546
Jiang, X., Zhou, T., Bai, R., Xie, Y.: Hydroxypyridinone-based iron chelators with broad-ranging biological activities. J. Med. Chem. 63, 14470–14501 (2020). https://doi.org/10.1021/acs.jmedchem.0c01480
Burgess, J., Rangel, M.: Hydroxypyranones, hydroxypyridinones, and their complexes. In: Advances in Inorganic Chemistry, pp. 167–243. Elsevier, Amsterdam (2008)
Saeedi, M., Eslamifar, M., Khezri, K.: Kojic acid applications in cosmetic and pharmaceutical preparations. Biomed. Pharmacother. 110, 582–593 (2019). https://doi.org/10.1016/j.biopha.2018.12.006
Baláž, Š, Uher, M., Brtko, J., Veverka, M., Bransová, J., Dobias, J., Pódová, M., Buchvald, J.: Relationship between antifungal activity and hydrophobicity of kojic acid derivatives. Folia Microbiol. 38, 387–391 (1993). https://doi.org/10.1007/BF02898762
Bransová, J., Brtko, J., Uher, M., Novotný, L.: Antileukemic activity of 4-pyranone derivatives. Int. J. Biochem. Cell Biol. 27, 701–706 (1995). https://doi.org/10.1016/1357-2725(95)00031-J
Loomis, L.D., Raymond, K.N.: Solution equilibria of enterobactin and metal-enterobactin complexes. Inorg. Chem. 30, 906–911 (1991). https://doi.org/10.1021/ic00005a008
Raymond, K.N., Cass, M.E., Evans, S.L.: Metal sequestering agents in bioinorganic chemistry: enterobactin mediated iron transport in E. coli and biomimetic applications. Pure Appl. Chem. 59, 771–778 (1987). https://doi.org/10.1351/pac198759060771
Rodgers, S.J., Lee, C.W., Ng, C.Y., Raymond, K.N.: Ferric ion sequestering agents. 15. Synthesis, solution chemistry, and electrochemistry of a new cationic analog of enterobactin. Inorg. Chem. 26, 1622–1625 (1987). https://doi.org/10.1021/ic00257a030
Hou, Z., Stack, T.D.P., Sunderland, C.J., Raymond, K.N.: Enhanced iron(III) chelation through ligand predisposition: syntheses, structures and stability of tris-catecholate enterobactin analogs. Inorg. Chim. Acta 263, 341–355 (1997). https://doi.org/10.1016/S0020-1693(97)05664-8
Tor, Y., Libman, J., Shanzer, A., Felder, C.E., Lifson, S.: Chiral siderophore analogs: enterobactin. J. Am. Chem. Soc. 114, 6661–6671 (1992). https://doi.org/10.1021/ja00043a008
Garrett, T.M., McMurry, T.J., Hosseini, M.W., Reyes, Z.E., Hahn, F.E., Raymond, K.N.: Ferric ion sequestering agents. 22. Synthesis and characterization of macrobicyclic iron(III) sequestering agents. J. Am. Chem. Soc. 113, 2965–2977 (1991). https://doi.org/10.1021/ja00008a027
Thakur, M., Baral, M., Kanungo, B.K.: Experimental and theoretical studies on coordination of a flexible cyclohexane based C3-symmetric tripodal ligand with iron, aluminum and gallium. Polyhedron 229, 116191 (2023). https://doi.org/10.1016/j.poly.2022.116191
Ng, C.Y., Rodgers, S.J., Raymond, K.N.: Ferric ion sequestering agents. 21. Synthesis and spectrophotometric and potentiometric evaluation of trihydroxamate analogs of ferrichrome. Inorg. Chem. 28, 2062–2066 (1989). https://doi.org/10.1021/ic00310a011
Thakur, M., Baral, M., Kanungo, B.K.: Design, synthesis, spectroscopic, photophysical and computational studies of a C3-symmetric hydroxyquinoline based tripod: TREN2OX and its interaction with Fe(III) and Al(III). J. Mol. Struct. 1248, 131436 (2022). https://doi.org/10.1016/j.molstruc.2021.131436
Sharma, S., Baral, M., Dash, D., Kanungo, B.K.: Synthesis, solution studies and DFT investigation of a tripodal ligand with 3-hydroxypyran-4-one scaffold. J. Incl. Phenom. Macrocycl. Chem. 101, 275–289 (2021). https://doi.org/10.1007/s10847-021-01088-0
Harrington, J.M., Chittamuru, S., Dhungana, S., Jacobs, H.K., Gopalan, A.S., Crumbliss, A.L.: Synthesis and Iron sequestration equilibria of novel exocyclic 3-hydroxy-2-pyridinone donor group siderophore mimics. Inorg. Chem. 49, 8208–8221 (2010). https://doi.org/10.1021/ic902595c
Xu, J., O’Sullivan, B., Raymond, K.N.: Hexadentate hydroxypyridonate iron chelators based on TREN-Me-3,2-HOPO: variation of cap size 1. Inorg. Chem. 41, 6731–6742 (2002). https://doi.org/10.1021/ic025610+
Liu, X., Xia, W., Jiang, Q., Xu, Y., Yu, P.: Synthesis, characterization, and antimicrobial activity of kojic acid grafted chitosan oligosaccharide. J. Agric. Food Chem. 62, 297–303 (2014). https://doi.org/10.1021/jf404026f
Xu, B., Kong, X.-L., Zhou, T., Qiu, D.-H., Chen, Y.-L., Liu, M.-S., Yang, R.-H., Hider, R.C.: Synthesis, iron(III)-binding affinity and in vitro evaluation of 3-hydroxypyridin-4-one hexadentate ligands as potential antimicrobial agents. Bioorg. Med. Chem. Lett. 21, 6376–6380 (2011). https://doi.org/10.1016/j.bmcl.2011.08.097
Dash, D., Baral, M., Kanungo, B.K.: Development of a flexible tripodal hydroxypyridinone ligand with cyclohexane framework: complexation, solution thermodynamics spectroscopic and DFT studies. ChemistrySelect. 6, 12165–12181 (2021). https://doi.org/10.1002/slct.202102962
Dash, D., Baral, M., Kanungo, B.K.: Synthesis of a new tetradentate chelator with 1-Hydoroxy-2(1H)-pyridinone (HOPO) as chelating unit: Interaction with Fe (III), solution thermodynamics and DFT studies. J. Mol. Struct. 1222, 128796 (2020). https://doi.org/10.1016/j.molstruc.2020.128796
Toni, M., Massimino, M.L., De Mario, A., Angiulli, E., Spisni, E.: Metal dyshomeostasis and their pathological role in prion and prion-like diseases: the basis for a nutritional approach. Front. Neurosci. (2017). https://doi.org/10.3389/fnins.2017.00003
Fouani, L., Menezes, S.V., Paulson, M., Richardson, D.R., Kovacevic, Z.: Metals and metastasis: exploiting the role of metals in cancer metastasis to develop novel anti-metastatic agents. Pharmacol. Res. 115, 275–287 (2017). https://doi.org/10.1016/j.phrs.2016.12.001
Mocchegiani, E., Giacconi, R., Malavolta, M.: Zinc signalling and subcellular distribution: emerging targets in type 2 diabetes. Trends Mol. Med. 14, 419–428 (2008). https://doi.org/10.1016/j.molmed.2008.08.002
Martindale, W., Reynolds, J.E.F.: The extra pharmacopoeia. Royal Pharmaceutical Press, London (1996)
Gemma, S., Kukreja, G., Campiani, G., Butini, S., Bernetti, M., Joshi, B.P., Savini, L., Basilico, N., Taramelli, D., Yardley, V., Bertamino, A., Novellino, E., Persico, M., Catalanotti, B., Fattorusso, C.: Development of piperazine-tethered heterodimers as potent antimalarials against chloroquine-resistant P. falciparum strains. Synthesis and molecular modeling. Bioorg. Med. Chem. Lett. 17, 3535–3539 (2007). https://doi.org/10.1016/j.bmcl.2007.04.077
Santos, M.A., Chand, K., Chaves, S.: Recent progress in multifunctional metal chelators as potential drugs for Alzheimer’s disease. Coord. Chem. Rev. 327–328, 287–303 (2016). https://doi.org/10.1016/j.ccr.2016.04.013
Crisponi, G., Nurchi, V.M.: Metal Ion Toxicity. In: Scott, R.A. (ed.) Encyclopedia of Inorganic and Bioinorganic Chemistry, pp. 1–14. Wiley, Hoboken (2015)
Andersen, O.: Principles and recent developments in chelation treatment of metal intoxication. Chem. Rev. 99, 2683–2710 (1999). https://doi.org/10.1021/cr980453a
Basketter, D.A., Angelini, G., Ingber, A., Kern, P.S., Menné, T.: Nickel, chromium and cobalt in consumer products: revisiting safe levels in the new millennium. Contact Dermatitis 49, 1–7 (2003). https://doi.org/10.1111/j.0105-1873.2003.00149.x
World Health Organization, International Agency for Research on Cancer, IARC Working Group on the Evaluation of Carcinogenic Risks to Humans eds: Cobalt in hard metals and cobalt sulfate, gallium arsenide, indium phosphide, and vanadium pentoxide. International Agency for Research on Cancer ; Distributed by WHO Press, Lyon, France : Geneva, Switzerland (2006)
Sunderman, F.W., Decsy, M.I., McNeely, M.D.: Nickel metabolism in health and disease. Ann. NY Acad. Sci. 199, 300–312 (1972). https://doi.org/10.1111/j.1749-6632.1972.tb46465.x
Angelova, M.G., Petkova-Marinova, T.V., Pogorielov, M.V., Loboda, A.N., Nedkova-Kolarova, V.N., Bozhinova, A.N.: Trace element status (Iron, Zinc, Copper, Chromium, Cobalt, and Nickel) in iron-deficiency anaemia of children under 3 years. Anemia 2014, 1–8 (2014). https://doi.org/10.1155/2014/718089
Nielsen, F.H., Shuler, T.R., McLeod, T.G., Zimmerman, T.J.: Nickel influences iron metabolism through physiologic, pharmacologic and toxicologic mechanisms in the rat. J. Nutr. 114, 1280–1288 (1984). https://doi.org/10.1093/jn/114.7.1280
Zhang, Y., Rodionov, D.A., Gelfand, M.S., Gladyshev, V.N.: Comparative genomic analyses of nickel, cobalt and vitamin B12 utilization. BMC Genomics 10, 78 (2009). https://doi.org/10.1186/1471-2164-10-78
Denkhaus, E., Salnikow, K.: Nickel essentiality, toxicity, and carcinogenicity. Crit Rev OncolHematol. 42, 35–56 (2002). https://doi.org/10.1016/s1040-8428(01)00214-1
Gans, P., Sabatini, A., Vacca, A.: Investigation of equilibria in solution. Determination of equilibrium constants with the HYPERQUAD suite of programs. Talanta 43, 1739–1753 (1996). https://doi.org/10.1016/0039-9140(96)01958-3
Alderighi, L., Gans, P., Ienco, A., Peters, D., Sabatini, A., Vacca, A.: Hyperquad simulation and speciation (HySS): a utility program for the investigation of equilibria involving soluble and partially soluble species. Coord. Chem. Rev. 184, 311–318 (1999). https://doi.org/10.1016/S0010-8545(98)00260-4
Irving, H., Williams, R.J.P.: 637. The stability of transition-metal complexes. J. Chem. Soc. (1953). https://doi.org/10.1039/jr9530003192
Nurchi, V.M., Crisponi, G., Lachowicz, J.I., de Jaraquemada-PelaezGuadalupe, M., Bretti, C., Peana, M., Medici, S., Zoroddu, M.A.: Equilibrium studies of new bis-hydroxypyrone derivatives with Fe3+, Al3+, Cu2+ and Zn2+. J InorgBiochem. 189, 103–114 (2018). https://doi.org/10.1016/j.jinorgbio.2018.09.013
Nurchi, V.M., de Guadalupe Jaraquemada-Pelaez, M., Crisponi, G., Lachowicz, J.I., Cappai, R., Gano, L., Santos, M.A., Melchior, A., Tolazzi, M., Peana, M., Medici, S., Zoroddu, M.A.: A new tripodal kojic acid derivative for iron sequestration: Synthesis, protonation, complex formation studies with Fe3+, Al3+, Cu2+ and Zn2+, and in vivo bioassays. J InorgBiochem. 193, 152–165 (2019). https://doi.org/10.1016/j.jinorgbio.2019.01.012
Nurchi, V.M., Crisponi, G., Arca, M., Crespo-Alonso, M., Lachowicz, J.I., Mansoori, D., Toso, L., Pichiri, G., Amelia Santos, M., Marques, S.M., Niclós-Gutiérrez, J., González-Pérez, J.M., Domínguez-Martín, A., Choquesillo-Lazarte, D., Szewczuk, Z., Antonietta Zoroddu, M., Peana, M.: A new bis-3-hydroxy-4-pyrone as a potential therapeutic iron chelating agent. Effect of connecting and side chains on the complex structures and metal ion selectivity. J InorgBiochem. 141, 132–143 (2014). https://doi.org/10.1016/j.jinorgbio.2014.09.002
Kohn, W., Sham, L.J.: Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133–A1138 (1965). https://doi.org/10.1103/PhysRev.140.A1133
Becke, A.D.: Density-functional thermochemistry. V. Systematic optimization of exchange-correlation functionals. J. Chem. Phys. 107, 8554–8560 (1997). https://doi.org/10.1063/1.475007
Bauernschmitt, R., Ahlrichs, R.: Stability analysis for solutions of the closed shell Kohn-Sham equation. J. Chem. Phys. 104, 9047–9052 (1996). https://doi.org/10.1063/1.471637
Pearson, R.G.: Chemical Hardness. Wiley, Hoboken (1997)
Pearson, R.G.: Absolute electronegativity and hardness correlated with molecular orbital theory. Proc. Natl. Acad. Sci. U.S.A. 83, 8440–8441 (1986). https://doi.org/10.1073/pnas.83.22.8440
Pearson, R.G.: Recent advances in the concept of hard and soft acids and bases. J. Chem. Educ. 64, 561 (1987). https://doi.org/10.1021/ed064p561
Koopmans, T.: Über die Zuordnung von Wellenfunktionen und Eigenwertenzu den EinzelnenElektronenEines Atoms. Physica 1, 104–113 (1934). https://doi.org/10.1016/S0031-8914(34)90011-2
Gans, P., Sabatini, A., Vacca, A.: Determination of equilibrium constants from spectrophometric data obtained from solutions of known pH: the program pHab. Anal. Chim. 89, 45–49 (1999)
TeVelde, G., Bickelhaupt, F.M., Baerends, E.J., Fonseca Guerra, C., Van Gisbergen, S.J.A., Snijders, J.G., Ziegler, T.: Chemistry with ADF. J. Comput. Chem. 22, 931–967 (2001). https://doi.org/10.1002/jcc.1056
Author information
Authors and Affiliations
Contributions
Shalini Singh: Experimental work, drafting; Minati Baral: Concept, supervision, and manuscript correction; B K Kanungo: validation and correction.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Singh, S., Kanungo, B.K. & Baral, M. Coordination competency of a flexible polyfunctional tripodal framework: an insight on solution thermodynamics and DFT studies. J Incl Phenom Macrocycl Chem 104, 219–231 (2024). https://doi.org/10.1007/s10847-024-01229-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10847-024-01229-1