
Abstract
Peptidoglycan, an essential component of the bacterial cell wall plays a critical role in protecting bacteria against osmotic lysis. The ATP-dependent MurC-F ligases are crucial for the early stages of peptidoglycan biosynthesis. MurE ligase is third in the series and catalyzes the addition of l-Lysine (l-Lys) in Gram-positive bacteria or meso-diaminopimelic acid (meso-A2pm) in most Gram-negative bacteria to form UDP-N-acetylmuramoyl-l-Ala-d-Glu-l-Lys/A2pm. The high substrate specific for l-Lys or meso-A2pm makes this enzyme an attractive target for the development of antibacterial agents. Several MurE inhibitors have been reported including phosphinates, peptidosulfonamides, napthylfuran-2-ones, benzene-1,3-dicarboxylic acids, phosphorylatedhydroxyethylamines, natural compounds, 5-benzylidenethiazolidin-4-ones, N-alkyl-2-alkynyl-4(1H)-quinolones, rhodanine substituted d-glutamic acids, 2,5-dimethyl pyrroles, 2,5-disubstitued furans, tetrahydroisoquinolines etc. In the present review we present an update status and structural information of MurE enzyme inhibitors which may be utilized for the design of potent inhibitors against this enzyme.
Similar content being viewed by others

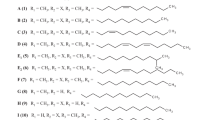

Avoid common mistakes on your manuscript.
Introduction
Resistance to antimicrobials agent among the pathogenic bacteria has emerged as a global threat in the past 20 years and has increased in the past decade [1]. The conserved underexploited antibacterial targets has enabled a search for the advent of genomics era especially in the early stage of peptidoglycan synthesis [2]. Peptidoglycan of the bacterial cell wall is a linear polymer of sugars cross-linked by short peptide bridges and plays a critical role in protecting bacteria against osmotic lysis [3]. A series of Mur enzymes (MurC-F) ensures the assembly of the pentapeptide part of the monomer unit. Mur ligases constitute a family of enzymes with common mechanistic and structural features [4]. In spherical Gram-positive bacteria such as the Streptococci and Staphylococci l-lysine residue appears at the third position of the cell wall peptide moiety (UDP-N-acetylmuramoyl-l-Ala-d-Glu-l-Lys), whereas in most Gram-negative bacteria such as Escherichia coli and Bacillus subtilis meso-diaminopimelic acid (meso-A2pm) appears at this position (UDP-N-acetylmuramoyl-l-Ala-d-Glu-meso-A2pm). Although l-Ala, l-Glu and l-homoserine has also been identified at these position in other bacterial species. MurE enzymes have been shown to discriminate between l-Lys and meso-A2pm and acts as a gatekeeper to ensure that only the specific substrate is incorporated in the peptidoglycan precursor. However, this difference in specificity reveals no significant difference in the protein sequence as only 28 and 32% identity is observed between the E. coli and S. pneumoniae or E. coli and B. subtilis sequences, respectively [3, 5]. Through their ε-amino group, meso-A2pm and l-Lys establish cross-linkage between the peptide units linked to the glycan chains. Hence they have a pivotal role in establishing cell shape and the integrity of the bacterial cell [6, 7].
The substrate and product of the MurE reaction are also known to be key components in the eukaryotic innate immunity machinery [8]. The E. coli MurE crystal structure in the presence of its product, UDP-N-acetyl-muramyl-tripeptide [3] showed three distinct globular domains of which two domains have similar topology as observed in the equivalent domain found in MurD three-dimensional structure. Domain 1 comprises residues 1–88 made up of two α-helixes which are surrounded to a five-stranded β-sheet. In Domain 2 seven α-helixes surrounded to a six-stranded central β-sheet, having similarity with MurD central domain and consists 90–338 residues. Whereas in domain 3 five α-helixes is surrounded to a six-stranded β-sheet made up with five parallel and one anti parallel strand and consists of 340–497 residues [9]. Across the four enzymes, domains 2 and 3 have conserved three-dimensional structure and similar topology. The Domain 2 also known as adenosine triphosphate (ATP) domain exhibit the highest level of structural similarity and sequence identities ranging from 22 to 26% [10]. This domain consists of essential structural motifs including the P-loop (motif 1), along with glutamate (motif 2) and histidine (motif 3) residues which are crucial for coordination with two magnesium ions and adenine specificity pocket (motifs 3 and 5). Among four Mur-ligases the P-loop (motif 1) is conserved where MurE consensus sequence is TGTNGKTTTT and residues are invariant across known MurE sequences [11]. Further, site-directed mutagenesis and chemical rescue experiments reveled an invariant carbamate-lysine derivative (lys-198) associated with MurD, -E, -F presumably required for Mg2+ binding and acyl phosphate formation [12].
As established with other Mur ligases, the MurE reaction proceeds by phosphorylation of the carboxyl group of the nucleotide precursor by ATP to form an acyl phosphate intermediate. The acyl phosphate is then attacked by the α-amino group of meso-A2pm to produce product, ADP and inorganic phosphate [13, 14]. In some of the bacterial species tetrahydrodipicolinic acid is directly converted into meso-A2pm by a dehydrogenase. In the peptidoglycan pathway, MurE catalyzes the addition of this meso-A2pm to UDP-MurNAc-dipeptide to yield UDP-MurNAc-tripeptide [15,16,17]. Absence of A2pm pathway in mammals instigated interest in the design and synthesis of A2pm analogues as antibacterial agents [18, 19]. In this review we report we present the most significant examples of MurE inhibitors that exhibit antibacterial activities reported in literature.
MurE inhibitors
The first transition-state analogue inhibitors of MurE ligases were phosphinate substituted dipeptides linked to the uridinediphosphate by hydrophobic spacer and displayed a tetrahedral geometry [20,21,22]. The IC50 of compound 1 (Fig. 1) (700 ± 50 µM) supports that MurE reaction follows a similar mechanism to that of other ATP-dependent amino acid ligases enzyme where the compound with structural similarity with the corresponding tetrahedral intermediate will bind tightly with the enzyme [23]. These works provide a starting point for the rational design of even more potent inhibitors of MurE [24].

Chemical structure of phosphinate derivative 1 and diphenylpeptidosulfonamide 2 as S. aureus MurE transition state inhibitor
In the search of new potent MurD inhibitor a series of peptidosulfonamides were synthesized as transition-state analogue, which turned out to be better inhibitor of MurE [25]. In fact the biphenyl derivative 2 (Fig. 1) exhibited inhibition of E. coli MurE as substrate analog with the IC50 value of 181 ± 18 µM. The reason for the poor inhibitory activity of peptidosulfonamides against E. coli MurE may be due to the elongation of the pseudo peptide backbone caused by the insertion of the additional methylene group, which may disrupt the active conformation of the molecule. Additional information from molecular modelling data has shown that sulfonamido group forms a weaker coordination bond with Mn2+, which might be the other reason for the inactivity of these compounds.
Napthylfuran-2-ones has been prepared and screened to identify multiple inhibitors of MurA-F enzymes [26]. This strategy of multi-target hypothesis was to prevent the development of drug resistance. In this series the compounds has been tested against MurB, C, D, E of both Staphylococcus aureus and E. coli and MurA of E. coli. Among them, compound 3 and 4 (Fig. 2) exhibited inhibitory activity against S. aureus (IC50 55 and 65 µM, respectively) and E. coli (IC50 13 and 16 µM, respectively) MurE enzymes. Compounds 6 and 7 exhibited lower activity against both S. aureus (IC50 > 64 in both cases) and E. coli (IC50 > 69 µM in both cases) MurE enzymes. On the other hand compound 5 showed activity against E. coli MurE with IC50 value of > 72 µM. These compounds are also evaluated in vitro against S. aureus and E. coli strains. Compound 4 which showed lowest IC50 value also exhibited lowest MIC value of 1–2 µg/mL against S. aureus and 2 µg/mL against E. coli. Whereas compounds 3, 5, 6 and 7 displayed promising MIC value against S. aureus (8, 8–16, 8 and 16–32 µg/mL, respectively) and E. coli (8, 16, 16, 8 µg/mL, respectively).

Chemical structures of napthyltetronic acid 3–7 as inhibitors of S. aureus and E. coli MurE ligase
Benzene-1,3-dicarboxylic acids were identified as MurD and MurE ligase inhibitors by pharmacophore-based virtual screening [27]. From this virtual screening approach a series of rigid analogues have been identified and tested in vitro against E. coli MurD and MurE enzymes. Among these hits only 2,5-dimethyl pyrrole analogue 11 (Fig. 3) exhibited inhibitory activity against E. coli MurD (IC50 270 µM) and MurE (IC50 32 µM) enzymes. Whereas furan analogue 12 displayed dual inhibitory activity against E. coli MurD (IC50 270 µM) and MurE (IC50 32 µM) enzymes, while compound 8 (IC50 79 µM), 9 (IC50 104 µM), 10 (IC50 97 µM) (Fig. 3) displayed moderate inhibitory activity against E. coli MurE.

Chemical structures of benzene-1, 3-dicaboxylic acid rigid analogues 8–12 as E. coli MurE ligase inhibitors
Further, phosphorylatedhydroxyethylamine derivatives have been developed to mimic transition-state analog for Mur ligase enzymes [28]. Among these compound 18 (Fig. 4) was the best inhibitor against S. aureus MurE with IC50 of 6 µM. Among the series electron donating group with resonance at para-position of phenyl ring increases the activity (compound 18), whereas electron withdrawing group at the same position decreases the inhibitory activity in compound 16 (S. aureus MurE IC50 57 µM) and 17 (S. aureus MurE IC50 120 µM). The electron withdrawing group at meta-position also decreased the activity in compound 15 (S. aureus MurE IC50 160 µM).

Chemical structure of phosphinate analogue 13–18 as MurE ligase inhibitors
From the Colombian plants species Ocotea macrophylla (Lauraceae), Dugandiodendron argyrotrichum (Magnoliaceae), Piper hispidum (Piperaceae), and Piper eriopodon (Piperaceae) natural compounds have been isolated and screen against M. tuberculosis MurE ligase inhibitory activity [29]. Among these 3-methoxynordomesticine hydrochloride (21, IC50 57 ± 14 µM) (Fig. 5) showed highest activity against M. tuberculosis MurE, whereas austrobailignan 6-threo and erythro diastereoisomers (20, IC50 286 ± 33 µM) exhibited lowest activity against this enzyme. Gibbilimbol-B (19, IC50 184 ± 16 µM), 3-methoxynordomesticine (21, IC50 67 ± 11 µM), N-methoxycarbonyl-3-methoxynordomesticine (22, IC50 75 ± 15 µM) displayed high to moderate M. tuberculosis MurE ligase activity. In vivo antibacterial screening of compound 21 against M. tuberculosis-ATCC 27294 and M. bovis ATCC 35734 showed MICs of 4 and 3 mg/L.

Chemical structures of naturally active compound 19–23 as M. tuberculosis MurE ligase inhibitors
For further development, 5-benzylidenethiazolidin-4-ones 24–28 (Fig. 6) has been designed with the aim to inhibit multiple Mur ligases [30]. It is observed that substituted rhodanine scaffold can offer several hydrogen bond interactions and this scaffold already has been reported act as MurD product mimics and also employed as phosphate mimetic or diphosphate surrogate. The ATP-binding and UDP-binding pockets are highly conserved for most Mur ligase enzymes. Based on these observations different derivatives has been designed and synthesized. 5-Benzylidenethiazolidin-2,4-dione 26 (IC50 3 µM) (Fig. 6) exhibited potent inhibitory activity against S. aureus MurE, whereas 5-benzylidenerhodanine 24 and 25 displayed inhibitory activity against S. aureus MurE (IC50 6 and 9 µM, respectively). N-substitution on the rhodanine ring resulted in compound 27 and 28 with slightly decreased MurE activity against S. aureus MurE (IC50 39 and 19 µM, respectively).

Chemical structures of 5-benzylidenethiazolidin-4-ones 24–28 as S. aureus MurE ligase inhibitors
In connection to the work on M. tuberculosis MurE inhibitors [31], five synthetic evocarpine-related quinolones has been screened against MurE of slow- and rapid-growing Mycobacterial species i.e. M. tuberculosis H37Rv, M. bovis BCG, M. smegmatis, M. bovis, M. fortuitum, M. phlei for further development [32]. Among all the screened compounds, compound 32 (Fig. 7) possessing cis-unsaturated aliphatic side chain showed highest activity against M. tuberculosis MurE with an IC50 value of 36 ± 16 µM. This compound also displayed inhibitory activity against M. tuberculosis ATCC 27294 (MIC 70.7 µM), M. bovis ATCC 35734 (MIC 28.3 µM), M. smegmatis ATCC 700084 (MIC 28.3 µM), M. fortuitum ATCC 6841 (MIC 1.41 µM) and M. phlei ATCC11758 (MIC 1.4 µM). Compounds 29–31 with trans-α,β-unsaturated double showed slight decreased MurE inhibitory activity along with decreasing length of the alkyl side chain 29 (IC50 52 ± 20 µM), 30 (IC50 72 ± 23 µM), and 31 (IC50 70 ± 25 µM).

Chemical structure of N-methyl-2-alkynyl-4-quinolones 29–33 and 34–39 as M. tuberculosis MurE ligase inhibitors
In a further development new N-alkyl-2-alkynyl-4(1H)-quinolones 34–39 (Fig. 7) has been synthesized and screened against M. tuberculosis MurE ligase [33]. All the compounds from this series showed inhibitory activity with IC50 values more than 200 µM, while compound 32 (Fig. 7) exhibited the lowest IC50 value of 200 µM. Further two more natural products have been reported from Hypericum acmosepalum species, namely Hypercalin B and HyperenoneA 40 (Fig. 8). Hypernone A showed an IC50 value of 320 µM against M. tuberculosis MurE.

Chemical structures of hyperenone-A 40 as M. tuberculosis and rhodanine substituted d-glutamic acid derivative 41 as S. aureus and E. coli MurE ligase Inhibitors
For the development of multi-target Mur ligases against Gram-positive and negative bacteria, rhodanine substituted d-glutamic acid derivative has been designed. As there was evidence that the thiazoledine-4-one act as weak multi-target inhibitor compared to the rhodanine moiety and as the product of MurD acts as a substrate in MurE. This evidence supports that residues important for d-glutamate acid binding in MurD has a same counterpart in MurE. From this hypothesis compound 41 (Fig. 8) is designed keeping rhodanine and d-glutamic acid constant whereas the linker between them is changed to more hydrophobic [34]. This resulted in activity against S. aureus MurE (IC50 17 ± 1.5 µM) and E. coli MurE (IC50 180 ± 60 µM) inhibitors with additional MurD inhibitory activity. This compound 41 was also tested against S. aureus-ATCC29213 (MIC 8 µg/ml) and MRSA-ATCC43300 (MIC 8 µg/mL).
In another development benzene-1,3-dicarboxilic acid possessing 2,5-dimethyl pyrrole nucleus [27] is further investigated because of their dual MurD/MurE inhibitory activity. From the virtual screening campaign benzene-1,3-dicarboxylic acid analogue has been identified as a conformational rigid mimetic of glutamic acid. Further modifications has been done by linking the 1,3 dicarboxylic with five member heterocyclic ring which was further linked with five or six member rings through methelene bridge [35]. The first series, where compound 11 has been identified as first dual MurD/MurE hits possess 2,5-dimethyl pyrrole moiety, where the methelene group was attached with 1-phenyl substituted dihydropyrimidine-4,6-dione and the other side was linked with 1,3-dicarboxylic acid. This hit has been optimized by placing various substitution at the benzene ring which is substituted with dihydropyrimidine-4,6-dione ring resulting in a series of active compounds. In this series compound 42 (IC50 330 µM), 43 (IC50 311 µM), 44 (IC50 330 µM) (Fig. 9) exhibited moderate inhibitory activity against E. coli MurE ligase. Next the atomic level mechanistic study has been performed using docking, molecular dynamics studies and the free energy calculation by Linear interaction energy (LIE) method. Results revealed that non-polar van der Waals interaction plays the major role in binding.

Chemical structure of 1,3-dicarboxylic acid 2,5-dimethylpyrrole derivative linked with dihydroprimidine-4,6-dione 42–44 and rhodadine moiety 45–50 as E. coli MurE ligase inhibitors
The encouraging result from the 1,3-dicarboxylic acid 2,5-dimethylpyrrole derivative has led the search of new series. From the previous series the dihydroprimidine-4,6-dione moiety (Fig. 9) from the parent scaffold has been replaced with rhodadine moiety [36]. With para-substitution has lend this series most successful one, where compound 45 (IC50 406 µM), 46 (IC50 494 µM), 47 (IC50 245 µM), 48 (IC50 303 µM), 49 (IC50 93 µM), 50 (IC50 89 µM) (Fig. 9) showed low to moderate inhibition of E. coli MurE ligase along with other MurC, D, F ligase inhibition. Among all compounds, 49 exhibited inhibition in low micro molar range (41–93 µM) against all four enzymes and also the steady-state kinetic study with MurD has been performed. In further modification dihydroprimidine-4,6-dione and rhodadine moiety attached to benzene ring has been replaced with other groups and 1,3-dicarboxylic acid from the other end has also replaced. But unfortunately none of the resultant compounds was found to be active except compound 51 (Fig. 10). Compound 51 exhibited activity against E. coli MurE ligase (IC50 44 µM) having dihydroprimidine-4,6-dione moiety.

Chemical structures of dihydropyridine-2,4-dione-1H-indole derivative 51 and 1,3-dicarboxylic acid-furan derivatives linked with rohdadine moiety 52–55 as E. coli MurE ligase inhibitors
Further investigation on furan based 1,3-dicarboxylic acid linked with rhodadine derivative have been carried out and resulted in a novel series of MurC-E multiple inhibitors [36]. Among the synthesized compounds 52–56 belonging to the first series, 52 showed activity against E. coli MurC, D, E, F (MurE IC50 272 µM) in micro molar range, while compounds 53 (IC50 251 µM), 54 (IC50 147 µM), 55 (IC50 233 µM) displayed activity only against E. coli MurE. For further insight into the mechanistic aspect classical SAR analogues-based medicinal chemistry approach has been used as non-availability of structural information about the binding mode of discovered compound 52 hindered the most optimal way of optimization. From MD simulation data it is observed that one carboxylic group established interaction with the ATP-binding site of MurD. In this way the optimization of this series from di to mono-carboxylic acid has been used and resulted in compounds 56–59. This modification revealed potent activity of compounds 56 (IC50 10 µM), 57 (IC50 11 µM), 58 (IC50 16 µM), 59 (IC50 16 µM) against E. coli MurE, while a decrease in activity against other Mur ligases enzyme was observed (Fig. 11).

Chemical structure of mono-carboxylic acid-furan derivative linked with rhodadine moiety 56–59 as E. coli MurE inhibitors
In another report tetrahydroisoquinolines (THA) has been prepared synthetically for screening against M. tuberculosis of MurE [37]. Among the thirty nine tested compounds, thirteen compounds displayed activity against M.tuberculosis MurE. A Bischler-Napieralski-mediated synthesis to ortho-cyclized tetrahydroisoquinolines resulted in compounds 60–62 with moderate to good inhibitory activity against M. tuberculosis MurE (IC50s 300, < 111 and 165 µM, respectively) (Fig. 12). All synthesized compounds were tested in vitro against M. tuberculosis H37 Rv-ATCC 27294 and M. bovis BCG-ATCC 35734 and their MIC values have been reported. Compounds 63–71 were prepared by phosphate-mediated pictet-spengler condensation between phenylethylamine and aldehydes and showed activity against M. tuberculosis MurE. Among these compounds 63 (IC50 < 111 µM), 64 (IC50 > 1000 µM), 65 (IC50 148 µM), 66 (IC50 471 µM), 67 (IC50 < 111 µM), 68 (IC50 > 1000 µM), 69 (IC50 186 µM), 70 (IC50 237 µM), 71 (IC50 837 µM) (Fig. 12) exhibited activity against M. tuberculosis MurE ligase. The alkaloid (S)-leucoxine 72 (Fig. 12) isolated from the Colombian Lauraceae tree Rhodostemonodaphnecrenaticupula madrinan exhibited IC50 value of 820 µM when tested against M. tuberculosis MurE [37].

Chemical structures of synthesized tetrahydroisoquinolines 60–71 and natural tetrahydro-isoquinoline 72 as M. tuberculosis MurE inhibitors. Chemical structure of 4,6-bis-anilino-1H-pyrrolo[2,3-pyrimidine] derivative 73 as E. coli MurE inhibitors
As all Mur ligases contain ATP-binding site, targeting this domain makes them vulnerable for multi target inhibitor. Inhibitors 73–76 (Fig. 13) against E. coli MurC-F ligase have been identified by targeting the ATP-binding site with known kinase inhibitors [38]. Each identified compound represents different structural classes. Among all the tested compounds, 73 a 4,6-bis-anilino-1H-pyrrolo[2,3-pyrimidine] analogue showed inhibitory activity at the lowest concentration (IC50 58 µM). Phenoxypyrimidine 74 (IC50 139 µM) andaza-stilbene 76 (IC50 79 µM) (Fig. 13) displayed moderate inhibitory activity against E. coli MurE, while alkynyl pyrimidine 75 showed inhibitory activity against this enzyme at the highest concentration (IC50 157 µM). These compounds also inhibited other Mur ligases at micro molar concentration. The kinetic study of compound 76 is reported. The MIC values of these compounds against S. aureus ATCC 29213 and E. coli ATCC 25922 has also been determined.

Chemical structures of phenoxypyrimidine derivative 74, alkynyl pyrimidine 75 and aza-stilbene 76 as E. coli MurE inhibitors
Approaches for the design of MurE inhibitors
These are several strategies employed to design novel class of MurE inhibitors. MurE enzyme consists of three distinct globular domains: A UDP-binding N-terminal domain, The ATP-binding central domain and the d-glutamic acid binding C-terminal domain. At the beginning the catalytic mechanism was targeted by assuming that MurE reaction resembles with many of the well characterized ATP-dependent ligases and this led to the design of peptide based phosphinate inhibitor [25, 39,40,41,42,43,44,45,46,47,48,49,50,51,52,53]. Based on this assumption compounds were designed with dipeptide analog linked by a hydrophobic linker to uridinediphosphate. This paved the way for the rational design especially targeting the catalytic mechanism by mimicking the structure of d-glutamic acid substrate in tetrahedral transition state. This strategy helped in the development of some novel scaffolds like analogues of hydroxyethylphosphinate, peptide-sulfonamides and napthyltetronic acids. Then later on 1,3-dicarboxylic acids were shown as tetrahedral analogues of d-Glu by steady-state kinetic study, where 1,3-dicarboxylic acid was mimicking the d-glutamic acid structure. Following this strategy two more novel scaffold has been discovered. One of them is 1,3-dicarboxilic acid-2,5-dimethylpyrrole derivatives linked with dihydropyrimidine-2,4-dione or rhodanine moieties the other one is 1,3-dicarboxylic acid linked with furan moiety.
The next approach used was to design nucleotide analogues by blocking the UDP-binding domain. As the UDP-binding domain of all Mur-ligases have very conserve residues which makes them as multi-target inhibitor. 5-benzylidenethiazolidin-4-ones were the first of this kind reported. These compounds were active in micro molar range and interactions with the binding pocket residues were observed by flanking the UDP-binding site. Another strategy employed for the design of MurE ligase inhibitors was by targeting the ATP-binding domain which is having the highest structural and sequence identity [8, 54] and presenting the shortened version of the classical P-loop consensus [11, 17] sequences whose conformation differs from the classical ATP-binding loop. Moreover, this domain has no similarity with ATP-utilizing human enzyme. Recently in silico virtual screening approach with known kinase inhibitors has been employed targeting the ATP-binding pocket. From this strategy several E. coli MurE inhibitors have been identified [38].
Further by exploiting the conformational changes in Mur ligases after substrate binding to form a potent active site may also be employed to identify MurE inhibitors. Specifically, this strategy may be employed for the strain specific design of MurE inhibitors. By developing a compound which can trap the enzyme in ‘open’ in an active form or capture them in topologically compact state where substrate can no longer access their binding sites may also be one of the strategy for the development novel MurE inhibitors. However, inhibitor of this type has not been reported yet for Mur ligases [50]. The penetrations of synthetic compounds remain elusive [55] and one of the biggest challenges to deal with antibacterial therapy based on Mur ligases enzyme. This can alternatively be achieved by conjugating to a stable moiety which can improve uptake via active bacterial transport [56] mechanism or can be used in synergy with different permeabilizers of bacterial envelopes like polymyxin B and cationic peptides [56,57,58,59,60].
Conclusions
MurE is an ATP dependent enzyme, act by adding l-lysine for Gram-positive bacteria and meso-DAP for Gram-negative bacteria to growing peptidoglycan strand of UDP-MurNAc-d-Glu. As it is involved in the early stage of cell wall synthesis and plays a crucial role in differentiation of Gram positive and negative bacteria’s cell wall. Though it has specificity for both l-lysine and meso-DAP, depending on the availability. This features makes an attractive target for the development of novel antibacterial agents as inactivation of these enzyme will led in hindrance of cross-linking at the later stage of cell wall synthesis. Several scaffolds like phosphinates, peptidosulfonamides, napthylfuran-2-ones, benzene-1,3-dicarboxylic acids, phosphorylatedhydroxyethylamines, 5-benzylidenethiazolidin-4-ones, N-alkyl-2-alkynyl-4(1H)-quinolones, rhodanine substituted d-glutamic acids, 2,5-dimethyl pyrroles, 2,5-disubstitued furans and tetrahydroisoquinolines have been investigated as MurE inhibitors. But unfortunately none these inhibitors exhibited potent activity against both Gram-positive and Gram-negative bacteria. For combating bacterial resistance multi-target oriented antibacterial agent may solve the problem for long term as the resistance due to mutation would have to occur in many target genes in a single bacterial generation. For multi-target Mur ligase inhibitor can be designed by targeting the UDP or ATP-binding site or mimicking the tetrahedral intermediate structure or by trapping the enzyme in ‘open’ inactive form. The most difficult aspect of modelling and synthetic chemical optimization of identified MuE inhibitors is frequent lack of biological activity. This may be due to the permeability barrier and efflux capabilities of bacteria [61]. One more possibility for the lack of activity for most MurE inhibitors in vivo is related to the action of the Mur pathway where the active sites being inaccessible to inhibitors perhaps by cancelling the intermediates and acting as a multi enzyme complex [63].
Abbreviations
- ATP:
-
Adenosine triphosphate
- d-Glu:
-
d-Glutamic acid
- IC50 :
-
Half maximal inhibitory concentration
- l-Ala:
-
l-Alanine
- meso-A2pm:
-
meso-Diaminopimelic acid
- MIC:
-
Minimum inhibitory concentration
- MurE:
-
UDP-N-acetylmuramoyl-l-Ala:d-Glu ligase
- UDP:
-
Uridine-5′-diphosphate
- UDP-MurNAc:
-
UDP-N-acetylmuramic acid
References
Jacobs, M.R.: Worldwide trends in antimicrobial resistance among common respiratory tract pathogens in children. Pediatr. Infect. Dis. J. 22, 109–119 (2003)
Silver, L.L.: Novel inhibitors of bacterial cell wall synthesis. Curr. Opin. Microbiol. 6, 431–438 (2003)
Gordon, E., Flouret, B.: Crystal structure of UDP-N-acetylmuramoyl-l-alanyl-d-glutamate: meso-diaminopimelate ligase from Escherichia coli. J. Biol. Chem. 276, 10999–11006 (2001)
Boniface, A., Bouhss, A.: The MurE synthetase from Thermotoga maritima is endowed with an unusual d-lysine adding activity. J. Biol. Chem. 281, 15680–15686 (2006)
Triolo, T.A., Chabin, R.M., Pompliano, D.L.: Cloning, expression and characterization of the Streptococcus pyogenes murE gene encoding a UDP-MurNAc-l-alanyl-d-glutamate: l-lysine ligase. Enzym. Microb. Technol. 35, 300–308 (2004)
Mengin-Lecreulx, D., Falla, T., Blanot, D., van Heijenoort, J., Adams, D.J., Chopra, I.: Expression of the Staphylococcus aureus UDP-N-acetylmuramoyl-l-alanyld-glutamate:l-lysine ligase in Escherichia coli and effects on peptidoglycan biosynthesis and cell growth. J. Bacteriol. 181, 5909–5914 (1999)
Glauner, B., Holtje, J.V., Schwarz, U.: The composition of the murein of Escherichia coli. J. Boil. Chem. 263, 10088–10095 (1988)
Ruane, K.M., Lloyd, A.J.: Specificity determinants for lysine incorporation in Staphylococcus aureus peptidoglycan as revealed by the structure of a MurE enzyme ternary complex. J. Biol. Chem. 288, 33439–33448 (2013)
El-Zoeiby, A., Sanschagrin, F., Levesque, R.C.: Structure and function of the Mur enzymes: development of novel inhibitors. Mol. Microbiol. 47, 1–2 (2003)
Smith, C.A.: Structure, function and dynamics in the mur family of bacterial cell wall ligases. J. Mol. Biol. 362, 640–655 (2006)
Sheng, Y., Sun, X., Shen, Y., Bognar, A.L., Baker, E.N., Smith, C.A.: Structural and functional similarities in the ADP-forming amide bond ligase superfamily: implications for a substrate-induced conformational change in folylpolyglutamate synthetase. J. Mol. Biol. 302, 425–438 (2000)
Dementin, S., Bouhss, A., Auger, G., Parquet, C., Mengin-Lecreulx, D., Dideberg, O., van-Heijenoort, J., Blanot, D.: Evidence of a functional requirement for a carbamoylated lysine residue in MurD, MurE and MurF synthetases as established by chemical rescue experiment. Eur. J. Biochem. 268, 5800–5807 (2001)
Bouhss, A., Dementin, S., van Heijenoort, J., Parquet, C., Blanot, D.: Crystal structure of UDP-N-acetylmuramoyl-l-alanyl-d-glutamate: meso-diaminopimelate ligase from Escherichia coli. FEBS Lett. 453, 15–19 (1999)
Barreteau, H., Kovac, A., Boniface, A., Sova, M., Gobec, S., Blanot, D.: Cytoplasmic steps of peptidoglycan biosynthesis. FEMS Microbiol. Rev. 32, 168–207 (2008)
Van Falk, P.J., Ervin, K.M., Volk, K.S., Ho, H.T.: Biochemical evidence for the formation of a covalent acyl-phosphate linkage between UDP-N-acetylmuramate and ATP in the Escherichia coli UDP-N-acetylmuramate:l-alanine ligase-catalyzed reaction. Biochemistry 35, 1417–1422 (1996)
Perdih, A., Kotnik, M., Hodoscek, M., Solmajer, T.: Targeted molecular dynamics simulation studies of binding and conformational changes in E. coli MurD. Proteins 68, 243–254 (2007)
Bouhss, A., Dementin, S., van-Heijenoort, J., Parquet, C., Blanot, D.: MurC and MurD synthetases of peptidoglycan biosynthesis: borohydride trapping of acyl-phosphate intermediates. Methods Enzymol. 354, 189–196 (2002)
Williams, R.M., Fegley, G.J., Gallegos, R., Schaefer, F., Pruess, D.L.: Asymmetric syntheses of (2S,3S,6S), (2S,3S,6R)-, and (2R,3R,6S)-2,3-methano-2,6-diaminopimelic. Acids studies directed to the design of novel substrate-based inhibitors of l, l-diaminopimelate epimerase. Tetrahedron 52, 1149–1164 (1996)
Auger, G., van Heijenoort, J., Vederas, J.C., Blanot, D.: Effect of analogues of diaminopimelic acid on the meso-diaminopimelate-adding enzyme from Escherichia coli. FEBS Lett. 391, 171–174 (1996)
Longenecker, K.L., Stamper, G.F., Hajduk, P.J., Fry, E.H., Jakob, C.G., Harlan, J.E., Edalji, R., Bartley, D.M., Walter, K.A., Solomon, L.R., Holzman, T.F.: Structure of MurF from Streptococcus pneumoniae co-crystallized with a small molecule inhibitor exhibits interdomain closure. Protein Sci. 14, 3039–3047 (2005)
Tanner, M.E., Vaganay, S., van-Heijenoort, J., Blanot, D.: Phosphinate inhibitors of the d-glutamic acid-adding enzyme of peptidoglycan biosynthesis. J. Org. 61, 1756–1760 (1996)
Strancar, K., Blanot, D., Gobec, S.: Design, synthesis and structure–activity relationships of new phosphinate inhibitors of MurD. Bioorg. Med. Chem. Lett. 16, 343–348 (2006)
Hrast, M., Sosič, I., Šink, R., Gobec, S.: Inhibitors of the peptidoglycan biosynthesis enzymes MurA-F. Bioorg. Med. 55, 2–15 (2014)
Zeng, B., Wong, K.K., Pompliano, D.L., Reddy, S., Tanner, M.E.: A phosphinate inhibitor of the meso-diaminopimelic acid-adding enzyme (MurE) of peptidoglycan biosynthesis. J. Org. Chem. 63, 10081–10086 (1998)
Gegnas, L.D., Waddell, S.T., Chabin, R.M., Reddy, S., Wong, K.K.: Inhibitors of the bacterial cell wall biosynthesis enzyme Mur D. Bioorg. Med. Chem. Lett. 8, 1643–1648 (1998)
Humljan, J., Kotnik, M., Boniface, A., Šolmajer, T., Urleb, U., Blanot, D., Gobec, S.: A new approach towards peptidosulfonamides: synthesis of potential inhibitors of bacterial peptidoglycan biosynthesis enzymes MurD and MurE. Tetrahedron 62, 10980–10988 (2006)
Mansour, T.S., Caufield, C.E., Rasmussen, B., Chopra, R., Krishnamurthy, G., Morris, K.M., Svenson, K., Bard, J., Smeltzer, C., Naughton, S., Antane, S.: Naphthyltetronic acids as multi-target inhibitors of bacterial peptidoglycan biosynthesis. Chem. Med. Chem. 2, 1414–1417 (2007)
Perdih, A., Kovac, A., Wolber, G., Blanot, D., Gobec, S., Solmajer, T.: Discovery of novel benzene 1, 3-dicarboxylic acid inhibitors of bacterial MurD and MurE ligases by structure-based virtual screening approach. Bioorg. Med. Chem. Lett. 19, 2668–2673 (2009)
Sova, M., Kovac, A., Turk, S., Hrast, M., Blanot, D., Gobec, S.: Phosphorylated hydroxyethylamines as novel inhibitors of the bacterial cell wall biosynthesis enzymes MurC to MurF. Bioorg. Chem. 37, 217–222 (2009)
Guzman, J.D., Gupta, A., Evangelopoulos, D., Basavannacharya, C., Pabon, L.C., Plazas, E.A., Munoz, D.R., Delgado, W.A., Cuca, L.E., Ribon, W., Gibbons, S.: Anti-tubercular screening of natural products from Colombian plants: 3-methoxynordomesticine, an inhibitor of MurE ligase of Mycobacterium tuberculosis. J. Antimicrob. Chemother. 65, 2101–2107 (2010)
Tomasic, T., Zidar, N., Kovac, A., Turk, S., Simcic, M., Blanot, D., Muller-Premru, M., Filipic, M., Grdadolnik, S.G., Zega, A., Anderluh, M.: 5-Benzylidenethiazolidin-4-ones as multitarget inhibitors of bacterial Mur ligases. Chem. Med. Chem. 5, 286–295 (2010)
Guzman, J.D., Wube, A., Evangelopoulos, D., Gupta, A., Hüfner, A., Basavannacharya, C., Rahman, M.M., Thomaschitz, C., Bauer, R., McHugh, T.D., Nobeli, I.: Interaction of N-methyl-2-alkenyl-4-quinolones with ATP-dependent MurE ligase of Mycobacterium tuberculosis: antibacterial activity, molecular docking and inhibition kinetics. J. Antimicrob. Chemother. 66, 1766–1772 (2011)
Wube, A., Guzman, J.D., Hufner, A., Hochfellner, C., Blunder, M., Bauer, R., Gibbons, S., Bhakta, S., Bucar, F.: Synthesis and antibacterial evaluation of a new series of N-Alkyl-2-alkynyl/(E)-alkenyl-4-(1H)-quinolones. Molecules 17, 8217–8240 (2012)
Osman, K., Evangelopoulos, D., Basavannacharya, C., Gupta, A., McHugh, T.D., Bhakta, S., Gibbons, S.: An antibacterial from Hypericumacmosepalum inhibits ATP-dependent MurE ligase from Mycobacterium tuberculosis. Int. J. Antimicrob. 39, 124–129 (2012)
Tomasic, T., Sink, R., Zidar, N., Fic, A., Contreras-Martel, C., Dessen, A., Patin, D., Blanot, D., Müller-Premru, M., Gobec, S., Zega, A.: Dual inhibitor of MurD and MurE ligases from Escherichia coli and Staphylococcus aureus. ACS Med. Chem. Lett. 3, 626–630 (2012)
Perdih, A., Hrast, M., Barreteau, H., Gobec, S., Wolber, G., Solmajer, T.: Benzene-1, 3-dicarboxylic acid 2, 5-dimethylpyrrole derivatives as multiple inhibitors of bacterial Mur ligases (MurC–MurF). Bioorg. Med. Chem. 22, 4124–4134 (2014)
Perdih, A., Hrast, M., Pureber, K., Barreteau, H., Grdadolnik, S.G., Kocjan, D., Gobec, S., Solmajer, T., Wolber, G.: Furan-based benzene mono-and dicarboxylic acid derivatives as multiple inhibitors of the bacterial Mur ligases (MurC–MurF): experimental and computational characterization. J. Comput. Aided Mol. Des. 29, 541–560 (2015)
Guzman, J.D., Pesnot, T., Barrera, D.A., Davies, H.M., McMahon, E., Evangelopoulos, D., Mortazavi, P.N., Munshi, T., Maitra, A., Lamming, E.D., Angell, R.: Tetrahydroisoquinolines affect the whole-cell phenotype of Mycobacterium tuberculosis by inhibiting the ATP-dependent MurE ligase. J. Antimicrob. 70, 1691–1703 (2015)
Hrast, M., Rožman, K., Ogris, I., Skedelj, V., Patin, D., Sova, M., Barreteau, H., Gobec, S., Grdadolnik, S.G., Zega, A.: Evaluation of the published kinase inhibitor set to identify multiple inhibitors of bacterial ATP-dependent mur ligases. J. Enzym. Inhib. Med. Chem. 34, 1010–1017 (2019)
Meek, T.D., Johnson, K.A., Villafranca, J.J.: Escherichia coli glutamine synthetase. Determination of rate-limiting steps by rapid-quench and isotope partitioning experiments. Biochemistry 21, 2158–2167 (1982)
Meek, T.D., Villafranca, J.J.: Kinetic mechanism of Escherichia coli glutamine synthetase. Biochemistry 19, 5513–5519 (1980)
Midelfort, C.F., Rose, I.A.: A stereochemical method for detection of ATP terminal phosphate transfer in enzymatic reactions. Glutamine synthetase. J. Biol. Chem. 251, 5581–5587 (1976)
Shi, Y., Walsh, C.T.: Active-site mapping of Escherichia colid-Ala-d-Ala ligase by structure-based mutagenesis. Biochemistry 34, 2768–2776 (1995)
Fan, C., Moews, P.C., Walsh, C.T.: Vancomycin resistance: structure of d-alanine: d-alanine ligase at 2.3 Å resolution. Science 266, 439–443 (1994)
Wright, G.D., Walsh, C.T.: d-Alanyl-d-alanine ligases and the molecular mechanism of vancomycin resistance. Acc. Chem. Res. 25, 468–473 (1992)
Mullins, L.S., Zawadzke, L.E., Walsh, C.T., Raushel, F.M.: Kinetic evidence for the formation of d-alanyl phosphate in the mechanism of d-alanyl-d-alanine ligase. J. Biol. Chem. 265, 8993–8998 (1990)
Falk, P.J., Ervin, K.M., Volk, K.S., Ho, H.T.: Biochemical evidence for the formation of a covalent acyl-phosphate linkage between UDP-N-acetylmuramate and ATP in the Escherichia coli UDP-N-acetylmuramate: l-alanine ligase-catalyzed reaction. Biochemistry 35, 1417–1422 (1996)
Bouhss, A., Mengin-Lecreulx, D., Blanot, D., van-Heijenoort, J., Parquet, C.: Invariant amino acids in the Mur peptide synthetases of bacterial peptidoglycan synthesis and their modification by site-directed mutagenesis in the UDP-MurNAc: l-alanine ligase from Escherichia coli. Biochemistry 36, 11556–11563 (1997)
Vaganay, S., Tanner, M.E., van-Heijenoort, J.E., Blanot, D.: Study of the reaction mechanism of the d-glutamic acid-adding enzyme from Escherichia coli. Microb. Drug Resist. 2, 51–54 (1996)
Eveland, S.S., Pompliano, D.L., Anderson, M.S.: Conditionally lethal Escherichia coli murein mutants contain point defects that map to regions conserved among murein and folyl poly-γ-glutamate ligases: identification of a ligase superfamily. Biochemistry 36, 6223–6229 (1997)
Walsh, C., Bradley, M., Nadeau, K.: Molecular studies on trypanothione reductase, a target for antiparasitic drugs. Trends Biochem. 16, 305–309 (1991)
Katoh, M., Hiratake, J., Kato, H., Oda, J.I.: Mechanism-based inactivation of E. coli γ-glutamyl cysteine synthetase by phosphinic acid-and sulfoximine-based transition-state analogues. Bioorg. Med. Chem. Lett. 6, 1437–1442 (1996)
Hiratake, J., Kato, H., Oda, J.I.: Mechanism-based inactivation of glutathione synthetase by phosphinic acid transition-state analog. J. Am. Chem. Soc. 116, 12059–12060 (1994)
Kato, H., Tanaka, T., Yamaguchi, H., Hara, T., Nishioka, T., Katsube, Y., Oda, J.I.: Flexible loop that is novel catalytic machinery in a ligase. Atomic structure and function of the loopless glutathione synthetase. Biochemistry 33, 4995–4999 (1994)
Payne, D.J., Gwynn, M.N., Holmes, D.J., Pompliano, D.L.: Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 6, 29–40 (2007)
Lewis, K.: Platforms for antibiotic discovery. Nat. Rev. Drug Discov. 12, 371–387 (2013)
Silver, L.L.: Does the cell wall of bacteria remain a viable source of targets for novel antibiotics? Biochem. Pharmacol. 71, 996–1005 (2006)
Nikaido, H.: Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 67, 593–656 (2003)
Baum, E.Z., Crespo-Carbone, S.M., Klinger, A., Foleno, B.D., Turchi, I., Macielag, M., Bush, K.: A MurF inhibitor that disrupts cell wall biosynthesis in Escherichia coli. Antimicrob. Agents Chemother. 51, 4420–4426 (2007)
Baum, E.Z., Crespo-Carbone, S.M., Foleno, B.D., Simon, L.D., Guillemont, J., Macielag, M., Bush, K.: MurF inhibitors with antibacterial activity: effect on muropeptide levels. Antimicrob. Agents Chemother. 53, 3240–3247 (2009)
Kouidmi, I., Levesque, R.C., Paradis-Bleau, C.: The biology of Mur ligases as an antibacterial target. Mol. Microbiol. 94, 242–253 (2014)
Funding
We would like to thank the Science and Engineering Research Board (SERB), Government of India for the financial support (No. EMR/2016/002981).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Saha, N., Azam, M.A. MurE inhibitors as antibacterial agents: a review. J Incl Phenom Macrocycl Chem 98, 127–136 (2020). https://doi.org/10.1007/s10847-020-01018-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10847-020-01018-6