
Abstract
The Gibbs energy of extraction experiments of ionophoric calixarenes with alkali metals from the literature have been correlated with electronic parameters of their “monomeric” structures calculated by AM1, PM3 and PM7 algorithms: energies of molecular orbitals and charge densities of oxygen and nitrogen atoms. We observed general correlations of the extraction constants with the charge on the carbonyl oxygen and specific correlations with MO-energy using the covalent term of the Klopman–Salem equation. The correlations for calixarenes with 5 and 6 phenolic units were very poor, and we attributed to the structural flexibility, which allow different optimized conformations for metal binding.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The born of alkali metal complexation can be attributed to the synthesis of dibenzocrown-6 by Pedersen [1], whose structure of the potassium complex showed that the cation is coordinated to the six ether oxygen donors of the macrocycle [2].
Pedersen, Lehn e Cram shared the Nobel Prize of 1987 by their contribution for the understanding of the factors that rule the complexation of ions and neutral molecules, and from their work begun the Supramolecular Chemistry.
Cation complexation has applications in cation detection [3], recovery and transport [4], phase-transfer catalysis [5] and biomimetical models [6]. New macrocycles based on crown-ethers, aza-crown, tia-crown and calixarenes [7] have been synthesized to obtain receptors by modulation of the size and hard-soft character of the ionophore to achieve complementarity with the metal cation [8].
Calixarenes with carboxy-methyl group linked to the phenolic hydroxil groups are well known as alkali cation receptors [9] and derivatives with phenolic esters displayed a correlation of the constant of extraction with the Hammett’s parameter σ+ [10]. Modeling of calix[4]arenes at all conformations performed by Hay and coworkers showed that K+, Rb+ and Cs+ have increased preference by interaction with the π-system of the calixarene [11], and ab initio calculations using density functional theory (DFT) were successful to predict chemical shifts for analogous calixarenes [12].
Several works using the same framework showed that the increase in the cavity from calix[4] to calix[5] and calix[6] displaces the selectivity from sodium towards potassium and cesium, whereas the presence of sulfur and nitrogen groups in the pendant arms displaces the selectivity toward “softer” cations, like Ag+ and Hg+2.
However, there are few reports dealing with the electronic parameters related with complexation [13], mainly by the multitude of entropic and enthalpic effects [14] determining the magnitude of the complexation, and in addition, dynamic factors related with the induced-fit promoted by the cation cannot be evaluated by these parameters.
The complexation of calixarenes with metal ions has been measured by conductimetric, extraction and displacement of the picrate band at UV spectra by several research groups [15]. These methods access different phenomena, but reflect the differences between the energies of the metal cation surrounded by solvent molecules and the metal cation surrounded by the calix ligand.
In this manuscript, we investigated the role of electronic parameters related with the extraction of alkali cations by the calixarene receptors, in order to evaluate the relative importance of these parameters for metal complexation. Although there are other measurements of the association constants, we chose the extraction by the high amount of data for heterogeneous extraction, while the data of homogeneous complexation were measured in different solvents, which does not allow the comparison of values. For example, the review of De Namor [15] compiles the thermodynamics data of interaction of calixarene macrocycles in MeCN, BzCN, EtOH, MeOH reported on the sources, but we were not able to find ten values from the same set of experimental conditions.
Experimental
The extraction results for the calix ionophores were obtained from literature results, usually reported as extraction percentage of the alkaline metal picrate from an organic solution (CH2Cl2 or CHCl3) to aqueous phase (%ext.), and the constants of extraction were obtained considering the contribution of activity coefficients for the metal ion [16].
Quantum Chemical Calculations: the semi-empirical program package MOPAC 2012 (http://openmopac.net) was used in all calculations. For each compound, computations were carried out with AM1 [17], PM7 [18] and PM3 [19] parametrization. Input files were conveniently generated and before to processed the output files were visualized with software RasMol 2.7.1 The molecular structures obtained in this way were used in a configurationally interaction calculation to compute electron density on phenolic oxygen, carbonyl oxygen and HOMO energies. Plots were drawn using Microcal Origin®.
Results and discussion
At the present, one of the most studied class of ionophores is calixarenes, by the combination of synthetic accessibility and the ability of further functionalization. The first report on the ionophoric properties of calixarenes described the synthesis of ethoxy-carboxy-methane-calix[4]arene (1, R 1 = t-bu, X = OEt) in cone conformation and its extraction behavior of picrate alkali metals from water to chloroform, with selectivity to sodium ion. The structure of the sodium salt [20] show that the four OCH2C = O groups surround the metal cation, and these semi-rigid groups arms fill the coordination sphere of the sodium.
We selected the extraction data with the backbone carboxy-methane-phenoxy 1, appended with groups at para (mostly tert-butyl, accessible by the calix synthesis), and alkyl-, aryl-, alkoxy- and amide radicals linked to the methoxy-carboxy-calix[n]arene for analysis of electronic parameters common to alkaline cation complexation. The extraction constant was obtained considering the equilibrium (Scheme 1):
The activity of the metal was obtained from the product of concentration and the activity coefficient, obtained by the simplified equation for ionic strength, whose value for all solutions was γ = 0.8; the values of [calix.M+.pic−] were obtained from the extraction data and the free Gibbs energy calculated from ΔGext. = − RT ln Kext are shown in Table 1.
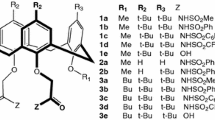
The constants of extraction were calculated from the extraction data of alkaline metals using calix[4]arene esters, ketones [21] and amides [22], [23] with the pendant group OCH2C(=O)–NR1R2. The latter have the same ionophoric properties towards alkali cations and also extract alkaline cations, and this behavior has been attributed due to the higher donicity of their carbonyl groups as compared to those of ester and ketone derivatives. Primary amines are not good ionophores by the internal hydrogen bonds between the N–H···O=C, which makes evident the role of the non-bonding pair of the oxygen atoms of carbonylic groups for the complexation [24]. The molecules based on calix[4]arene suitable for our study are shown in Scheme 2.

Basic structure of methoxy-carboxy-calix ionophores

Calix[4]arene ionophores used for modeling interactions
Ionic or covalent?
The complexation of the metal ion with calixarenes results from ionic and covalent effects, related with the interaction of the charge of the metal ion charge and the lone pair of the oxygen and/or nitrogen atoms of the ionophore.
In order to check the role of electronic parameter and macrocyclic effect, the calculations of electronic charge density and orbital energies charge densities were performed for the “monomeric” unit 2. The electronic features selected for search of correlations were the energy of EHOMO, EnC=O, En Ophe, qOcar, qOester/Namide and qOphe, as shown in Scheme 3.

Selected parameters related with ion metal interaction
We chose six parameters that can be related with the interaction metal ion–ionophore: EHOMO-related with covalent interaction with the aromatic ring; EnOphe related with covalent interaction with the phenolic oxygen; EnC=O related with the covalent interaction with the carbonyl oxygen; qOester,Namide related with electrostatic interaction with the ester oxygen or amide nitrogen—this interaction occurs when the metal ion is greater than the cavity formed by the OCH2C=O moieties and is located out of this cavity; qOcar related with the electrostatic interaction with the carbonyl oxygen and qOphe related with electrostatic interaction with the phenolic oxygen, and selected values are shown in Table 2.
The interaction of the metal can be rationalized by the resonance structures related with covalent (I and IV) and ionic (II and III) interaction between the calix ionophore and the ion metal shown in Scheme 4. Structures I and IV are related with covalent binding between metal and oxygen, whereas structures II and III are related with dipole-metal interaction. In addition, III and IV reflect the conjugation between the C=O and the non-ligand electron pair and increase the charge density over the carbonyl oxygen, which is important for amides, less important for esters and negligible for ketones. Structures like I and II are available to ketones, whereas for esters and amides all structures are available. This difference can lead to different relationships between the weight of the contribution of electrostatic or covalent binding.

Resonance structures for ion metal-ionophore complex
The equation of Klopman–Salem [26] provides an interpretation of a donor–acceptor electron pair, like nucleophile–electrophile or Lewis’ base-acid pair, where the ionophore is the base and the metal is the acid. The simplified form of the equation is shown below:
The first term is related with electrostatic interaction, mainly related with ionic character of the bond, whereas the second term is related with the superposition of the orbitals from the atoms, the covalent term. When the energies of the HOMO and LUMO becomes similar, this term becomes more important, and when the energies of HOMO and LUMO are more different, the ionic term prevails.
For calixarene ionophores, electrostatic interaction is related with charge density mainly over the carbonyl oxygen (qOcar), so if this kind of interaction has the main role for ΔGext., we expect to find correlation between ΔGext. and qOcar. Considering the values of cacid, cbase and β for the interaction of each metal ion with a serie of related calixarene ionophores as constants, the Klopman–Salem equation predicts a correlation between ΔGext and (EHOMO − ELUMO)−1 for orbital interaction.
At the first sight, we expect that the HOMO should be related with the energy of the lone pair of the carbonyl oxygen. However, the maps of electronic density of the orbitals for the monomer (Fig. 1) revealed that the HOMO for the monomeric unit of the ionophore is located on the aromatic ring, as well the HOMO-1, and the highest energy orbital which has an important electronic density on the lone pair of the carbonyl oxygen (EnC=O) is the HOMO-3 in most of the molecules, whereas the electronic density of HOMO-2 lies on the lone pair of the phenolic oxygen (EnOphe).

Draws of molecular orbitals of monomer 4 (R = OMe): a Orbital HOMO, b Orbital HOMO -1, c orbital HOMO-2, d orbital HOMO-3
The correlations of ΔGext. for each metal ion with (EHOMO − ELUMO)−1 can be used to evaluate the covalent interaction with aromatic ring, whereas (EnC=O − ELUMO)−1 is useful for the covalent interaction with the non-ligand pair of carbonyl oxygen and (EnOphe − ELUMO)−1 can be used to evaluate the covalent interaction with the phenolic oxygen.
Some questions related to the interaction of the receptor with the metal arise: (1) alkaline metals are complexed on the same way? (2) the macrocyclic effect adds a new energetic component of electronic nature? (3) what kind of interaction is more important: electrostatic or covalent interaction? (4) what is the interaction that promote selectivity?
Plots for metal ions
In order to give some basis to answer the first question pointed above, we thought that the plots between the values of ΔGext. for different alkaline metals could give us some correlations and the convergence of parameters for different metals. The other questions could be answered by correlations from ΔGext. and electronic parameters which could be related with the complexation, like the charge and energy of orbitals and the covalent term from Klopman–Salem equation.
To address the first question, we drew plots of ΔGext. between different alkaline metals from different ionophores (Fig. 2) and compare the values of the slopes (b) and correlation factors (R), which are listed in Table 3.

Plots for metal–metal values of ΔGext.: K+ × Li+ (left), K+ × Na+(center), Cs+ × Li+(right)
The plots with better correlations are, in general, those between ions of sequential rows of periodic table, except for Li+, whose best correlation was achieved with K+. Values of slopes near of the unity should be an indicative that the same factors rule. The plots of Rb+ and Cs+ with the other metals show a split in the graphic, with two parallel straight lines. This behavior comes from the nature of the ionophore, where the data from esters and ketones show an almost straight line (R = 0.97 − K+ × Cs+), whereas the data from amides are more dispersed (R = 0.82).
The plots of ΔGext. of all ion metals with qOphe and qOester/qNamide did not show correlation for all algorithms used, and the plots with EHOMO, EnC=O, qOcar, and (EnC=O − ELUMO)−1 showed some linearity for molecules with same organic function. Plots between electronic parameters did not correlate, that is good for our study because the differences in the association constants can be attributed to one of the parameters.
The ΔGext. for all metals decreases in the series ketones > esters > amides as a general trend, and the plots showed good to fair correlations with qOcar from the data generated by PM7 algorithm, although Cs+ exhibits the best correlation using PM3 instead PM7. The correlations for plots from Li+ (Fig. 3) and K+ were above R = 0.92 and for the other metals the values of R fell below 0.88. The correlation of the values using other parameters were very poor and no trends were detected. These results are consistent with a dependence of the charge on the oxygen, represented by structures II and III in Scheme 4.

Plots of −ΔGext. with −qOcar using AM1, PM3 and PM7 algorithms
The lost of quality for sodium may be related with the high complementarity with size cavity and ionic radius, and additional sterical factors have a major role, like the crowding between the t-butyl groups in the upper rim, which enforces the OCH2C=O moiety to stay closer in the space.
Potassium cation interacts with the aromatic π clouds [25], of residues of aromatic aminoacids (Phe, Tyr, Trp) in enzymes, and calixarenes fixed in 1,3-alternate conformation are selective ionophores for potassium [28]. However, we did not observe correlation with the parameters related with the energy of the aromatic orbitals (EHOMO, EHOMO−1) or the value of (EHOMO − ELUMO)−1.
Ions with bigger ionic ratio like Rb+ and Cs+ showed fair correlation with qOcar(R = 0.877 and 0.856, respectively), despite some have evidences of interaction with π-clouds in other calixarene-based receptors, but again, we did not detect correlation with parameters related with the energy of the π-system.
The search of correlations can be fine tuned using the extraction values for the same organic function, which indicates the parameter that differentiate the extraction behavior in the same basic structure. The plots between −ΔGext. for all metals with the electronic parameters for ionophoric esters were drawn, and the best plot for Li+ was achieved for the covalent Klopman–Salem parameter from the lone pairs of the phenolic oxygen (En Ophe − ELUMO)−1 (Fig. 4). The best correlation was obtained for Li+ (R = 0.997) and the worst was for K+ (R = 0.813), and for the other alkaline ions were good (R > 0.95).

Plots for the Gibbs energy of extraction of ionophore ester calix[4]arenes with Klopman–Salem parameter
To investigate the hypothesis of the ionic contribution for the enhanced ionophoric properties of amide-calixarenes. If it is true, we can anticipate that the main contribution of complexation should come from, and the related parameter is qOcar.
The plots of ΔGext. of amides from different metals (Fig. 5) showed again a different behavior for sodium, whereas lithium, potassium, rubidium e cesium have good to fair correlations.

Correlations for ionophore ester calix[4]amides
The plots with electronic parameters showed better correlations with Klopman–Salem covalent parameter instead of ionic, however the parameter which gave the best results was (EnC=O − ELUMO)−1 instead of (EOphe − ELUMO)−1. Our hypothesis about the main contribution of the lone pair of carbonyl oxygen for enhanced interaction of amides was right, but the best parameter was the covalent term of Klopman–Salem from PM7 algorithm.
The behavior for metals was the same than that observed for esters: fair correlation for Li+, K+, Rb+ and poorer for Na+ and Cs+. The best fit of the cavity for Na+ imposes some sterical restrictions that electronic parameters were not able to catch whereas Cs+ is a large cation, its charge is dispersed and the interaction is more diffuse.
The best plot for Cs+ was achieved using the covalent term with the energy of the orbital corresponding to the lone pair on the nitrogen atom (En−N – ELUMO)−1, and this is coherent with the position of the cation below the nitrogen atoms, interacting with the charge of the carbonyl groups and nitrogen lone pairs.
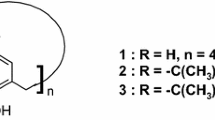
When the same data of electronic parameters are applied for the complexation of alkali metal cations with calix[5,6]arenes [21–27] (Scheme 5) with similar covalent structure [28], there is a complete lack of correlation, even if we use only the values for p-t-bu derivatives. The correlations using with all parameters for this set of data from calixarenes with 5 and 6 units were very poor, with the values of r in the range 0.3–0.7.

Calix[5] and calix[6]arene ionophores
We attribute the lack of correlation to the increase of the number of conformations by rotation around the methylene bridges, and the importance of the conformational equilibrium is more important than electronic parameters for these more flexible hosts. The increase of mobility of the phenolic moieties of calix[5] and [6]arenes allows that other conformations, instead the pure cone, be the most favorable to complex the cation. In this case, parameters related with packing of the groups around the cation is more important. The data showed that the ionophores with t-butyl present in the bottom arms are the best for complexation for all metals.
This proposal agrees with the 1H-NMR and X-ray diffraction results. For example, the pentamer (R 1 = X = t-bu) possesses a very distorted cone conformation in the solid state, and the pentamer (R 1 = H, X = t-bu) exists in a distorted 1,2-alternate conformation. Structural data of sodium and rubidium complexes of pentamer (R 1 = X = t-bu) show the cation deeply encapsulated by the ethereal and carbonyl oxygen atoms in distorted cone conformation.
Similar structures of calix[4]arenes complexed with Na+ show the cation surrounded by the eight oxygen atoms at cone conformation and C4v symmetry around the cation [29], but the aromatic rings have almost the same conformation than uncomplexed calix ionophores. The overall reorganization is lesser than for calix[5]arenes, so dynamic factors are less important, and electronic can display a major role.
Conclusion
Electronic parameters displays a major role for the complexation of Li+, Na+ and K+ with calix[4]arene derivatives functionalized with the carboxy-methoxy group, and in general, qOcar parameter has a major role for the magnitude of complexation, and implies that the electrostatic interaction is the main force responsible by the complexation.
The analysis of the results for each function shows that the term of the Klopmann–Salem’s equation related with covalent interaction of the LUMO of the metal ion with the orbital of phenolic oxygen correlates well for the ester derivatives, whereas for amides the best correlation for amides was achieved from the covalent term related with the energy of the non-bonding electron pair of the carbonyl oxygen, that implies to different secondary interaction for each function to give selectivity. The metal ion goes deeper in the pseudo-cavity formed by the OCH2C=O moiety for the ester derivative and interacts with the phenol oxygen atoms, whereas for amide derivatives, the secondary interaction proceeds with the electrons of the carbonyl oxygen.
For Rb+ and Cs+, the correlations have some conflicting results, which probably come from the different mode of binding of the cation. Calixarenes with 5 and 6 units have more conformations able to bind the metal cation and does not fit well with these electronic parameters.
References
Pedersen, C.J.: Cyclic polyethers and their complexes with metal salts. J. Am. Chem. Soc. 89(26), 7017–7036 (1967)
Blake, A.J., Gould, R.O., Parsons, S., Radek, C., Schröder, M.: Potassium dibenzo-18-crown-6 triiodide. Acta Cryst. C 52(1), 24–27 (1996)
Patra, S., Maity, R., Gunupuru, R., Aghihotri, P., Parimal, P.: Calixarenes: versatile molecules as molecular sensors for ion recognition study. J. Chem. Sci. 124(6), 1287–1299 (2012)
Ansar, S.A., Mohapatra, P.K., Iqbal, M., Kandwal, P., Huskins, J., Verboom, W.: Novel diglycolamide-functionalizad calix[4]arenes for actinide extraction and supported liquid membrane studies: role of substituents in the pendent arms and mass transfer modeling. J. Membr. Sci. 430, 304–311 (2013)
Simoes, J.B., da Silva, D.L., de Fatima, A., Fernandes, S.A.: Calix[n]arenes in action: useful host–guest catalysis in organic chemistry. Curr. Org. Chem. 16(8), 949–971 (2012)
Seneque, O., Rager, M.N., Giorgi, M., Reinaud, O.: Calix[6]arenes and zinc: novel biomimetic receptors for neutral molecules. J. Am. Chem. Soc. 122(26), 6183–6189 (2000)
Jacques, V., Harrowfield, J. (eds.): Calixarenes in the nanoworld. Springer, The Netherlands (2007)
Chinta, J.P., Ramanujam, B., Rao, C.P.: Structural aspects of the metal ion complexes of the conjugates of calix[4]arene: crystal structures and computational models. Coord. Chem. Rev. 256, 2762–2794 (2012)
Sliwa, W., Girek, T.: Calixarene complexes with metal ions. J. Incl. Phenom. Macrocycl. Chem. 66, 15–41 (2010)
Lazzarotto, M., Francisco, M.M., Nachtigall, F.F., Lazzarotto, M.: Substituent effects on ion complexation of para-tert-butylcalix[4]arene esters. J. Phys. Org. Chem. 19(11), 765–770 (2006)
Hay, B.P., Nicholas, J.B., Feller, D.: Novel binding modes in tetramethoxycalix[4]arene: implications for ligand design. J. Am. Chem. Soc. 122(41), 10083–10089 (2000)
Kara, I., Kart, H.H., Kolsuz, N., Karakus, Ö.Ö., Deligöz, H.: Ab initio studies of NMR chemical shifts for calix[4]arene and its derivatives. Struct. Chem. 20, 113–119 (2009)
Kumpf, R.A., Dougherty, D.A.: A mechanism for ion selectivity in potassium channels: computational studies of cation-pi interactions. Science 261(5129), 1708–1710 (1993)
Talanova, G.G., Talanov, V.X., Hwang, H.S., Park, C., Surowiec, K., Bartsch, R.A.: Rigid versus flexible: how important is ligand “preorganization” for metal ion recognition by lower rim-functionalized calix[4]arenes? Org. Biomol. Chem. 2(18), 2585–2592 (2004)
de Namor, A.F., Cleverley, R.M., Zapata-Ormachea, M.L.: Thermodynamics of calixarene chemistry. Chem. Rev. 98(7), 2495–2525 (1998)
Arduini, A.A., Reverberi, A., Ungaro, R., Andreetti, G.D., Ugozzoli, F.: The preparation and properties of a new lipophilic sodium selective ether ester ligand derived from p-t-butylcalix[4]arene. Tetrahedron 42(7), 2089–2100 (1986)
Dewar, M.J.S., Zoebisch, E.G., Healy, E.F., Stewart, J.J.P.: Development and use of quantum mechanical molecular models. 76. AM1: a new general purpose quantum mechanical molecular model. J. Am. Chem. Soc. 107(13), 3902–3909 (1985)
Stewart, J.J.P.: MOPAC 2012—Stewart computational chemistry, Colorado Spring. http://OpenMOPAC.net
Stewart, J.J.P.: Optimization of parameters for semiempirical methods I. Method. J. Comput. Chem. 10(2), 209–220 (1989)
de Namor, A.F., Kowalska, D., Castellano, E.E., Piro, O.E., Sueros Velarde, F.J., Salas, J.V.: Lower rim calix(4)arene ketone derivatives and their interaction with alkali metal cations. Structural and thermodynamic (solution and complexation) characterisation of the tetraphenyl ketone derivative and its sodium complex. Phys. Chem. Chem. Phys. 18, 4010–4021 (2001)
Arnaud-Neu, F., Collins, E.M., Deasy, M., Ferguson, G., Harris, S.J., Kaitner, G., Lough, A.J., McKervey, M.A., Marquis, E., Ruhl, B.L., Schwing-Weill, M.J., Seward, M.: Synthesis, X-ray crystal structures, and cation-binding properties of alkyl calixaryl esters and ketones, a new family of macrocyclic molecular receptors. J. Am. Chem. Soc. 111(23), 8681–8691 (1989)
Arnaud-Neu, F., Guerra, S., McGregor, W., Ziat, K., Schwing-Weill, M.J., Barrett, G., McKervey, M.A., Marrs, D., Seward, E.M.: Cation complexation by chemically modified calixarenes. Part 7. Transport of alkali cations by p-tert-butylcalix[n]arene esters and amides. J. Chem. Soc., Perkin Trans. 2, 113–118 (1995)
Arnaud-Neu, F., Barrett, G., Fanni, S., Marrs, D., McGregor, W., McKervey, M.A., Schwing-Weill, M.J., Vetrogon, V., Wechsler, S.: Extraction and solution thermodynamics of complexation of alkali and alkaline-earth cations by Calix[4]arene amides. J. Chem. Soc. Perkin Trans. 2, 453–461 (1995)
Arnaud-Neu, F., Barboso, S., Berny, F., Casnati, A., Muzet, N., Pinalli, A., Ungaro, R., Schwing-Weill, M.J., Wiipff, G.: Modulation of cation binding in calix[4]arene amides: synthesis, complexation and molecular modelling studies. J. Chem. Soc. Perkin Trans. 2, 1727–1738 (1999)
Klopman, G.: Chemical reactivity and the concept of Charge- and Frontier-controlled Reactions. J. Am. Chem. Soc. 90(2), 223–234 (1968)
Murayama, K., Katsuyuki, A.: Cation-π interactions between potassium ions and aromatic rings. Crystal structures of three potassium complexes of calix[6]arene. Inorg. Chim. Acta 281, 36–42 (1998)
Beer, P.D., Drew, M.G.B., Gale, P.A., Leeson, P.B., Ogden, M.I.: Structures of potassium encapsulated within the 1,3-alternate conformation of calix[4]arenes. J. Chem. Soc., Dalton Trans. 23, 3479–3485 (1994)
Bell, S.E., Browne, J.K., McKee, V., McKervey, M.A., Malone, J.F., O’Leary, M., Walker, A., Arnaud-Neu, F., Boulangeot, O., Mauprivez, O., Schwing-Weill, M.J.: Cation complexation by chemically modified calixarenes. 11. complexation and extraction of alkali cations by calix[5]- and -[6]arene ketones. crystal and molecular structures of calix[5]arene ketones and Na+ and Rb+ complexes. J. Org. Chem. 63(3), 489–501 (1998)
Ferguson, G., Kaitner, B., McKervey, M.A., Seward, E.M.: Synthesis, X-ray crystal structure, and cation transfer properties of a calix[4]arene tetraketone, a new versatile molecular receptor. J. Chem. Soc., Chem. Commun. 1987, 584–585 (1987)
Acknowledgments
The authors acknowledge to CNPq and INCT-Catalise for financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lazzarotto, M., Weinert, P.L. & Lazzarotto, M. Electronic parameters of cation complexation by calixarene ionophores. J Incl Phenom Macrocycl Chem 80, 313–322 (2014). https://doi.org/10.1007/s10847-014-0404-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10847-014-0404-8