
Abstract
The structural differences in the interaction between natural γ-cyclodextrin and bile salts common in rat, dog and man was were investigated by 1H-ROESY and 13C NMR and molecular modeling and the thermodynamic parameters of the reaction by isothermal titration calorimetry. The γ-cyclodextrin was selected based upon its frequent use in drug formulation as excipients to facilitate the solubilisation of drug substances with low aqueous solubility upon oral administration. The NMR studies and the molecular modeling demonstrated an interaction with inclusion of the C-ring of the steroid body of the bile salt and partly inclusion of the B and D ring. A large variation was observed in the stability constants among the investigated bile salt. The variations in the enthalpic and entropic contributions to the overall Gibbs free energy and consequently the stability constants revealed structural differences between the bile salts, where bile salts with a hydroxyl group on C12 has a weaker interaction than the bile salts without the hydroxyl group. Based upon the theoretical calculations of the available surface area the differences observed in the entropic contribution seems to be mainly driven by dehydration effects.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cyclodextrins (CDs) are cyclic macromolecules consisting of glucose units linked together with α-1,4-glycosidic bonds, which creates a shape of a truncated cone, with cavities of different size depending on the number of glycopyranose units. The most frequently investigated CDs consist of six, seven, or eight units of α-d-(+)-glucopyranose, denoted α, β, or γ-CDs, respectively [1]. CDs are capable of including smaller molecules into their cavity region by non-covalent interactions with the inside of the CD; primarily through hydrophobic interactions. In the pharmaceutical field this phenomenon is utilized to enhance the solubility of drugs, in order to enhance the bioavailability of compounds with low aqueous solubility administered both orally and parentally (reviewed by e.g. [1–8]).
The oral bioavailability of both α-, β-, and γCDs is very low [8], therefore only the free form of the drug, which is in equilibrium with the molecular complex, is available for absorption. For compounds with a high CD stability constant, the bioavailability of the compound may therefore be reduced if excess CD are coadministered [9]. The release of the compound from the complex is based upon simple dilution effects upon ingestion according to the law of mass action, but also displacement of the drug molecule from the CD cavity by compounds present within the gastrointestinal tract [10]. Of the compounds within the gastrointestinal tract the bile salts have been demonstrated to have an important role in the displacement from the CD cavity [11]. It is therefore of importance to understand the interaction between CDs and bile salts better to facilitate a better and more rational drug formulation process in oral formulations utilizing CDs.
CDs are α-1,4 oligomers of glucose and are therefore susceptible to hydrolysis by amylase. However, only the larger γCD is readily broken down enzymatically, possibly due to the high curvature of the smaller CDs [12, 13]. Modern drug design approaches are based on combinational chemistry and quantitative structure–activity relationship, which generates a vast numbers of new potent compounds, however, unfortunately often endowed with high molecular weights, high octanol/water partition coefficients (log P) and low water solubilities [14]. This possible enzymatic hydrolysis of γCD in the intestine combined with a larger cavity capable of complexing larger compounds when compared to the classically used βCDs makes γCDs of increasing interest for pharmaceutical applications. The interaction between some bile salts and γCD have been investigated by NMR [15, 16], affinity capilary electrophoresis [17], ITC [18], and phase solubility studies [19]. According to Alvaro et al. [20] six different bile salts dominate in the intestine of man, rat and dog. These are, taurocholate (TC), taurodeoxycholate (TDC), taurochenodeoxycholate (TCDC), glycocholate (GC), glycodeoxycholate (GDC) and glycochenodeoxycholate (GCDC), see Fig. 1, but the thermodynamics of binding to γCD have only been investigated for TC. The purpose of the current study was therefore to investigate the differences in binding thermodynamics and mode of binding of bile salts present in man, rat and dog to natural γCD by ITC, 1H-ROESY and 13C NMR and molecular modeling to increase the understanding of the interaction and to facilitate rational drug formulation with γCDs.

Chemical structure of investigate bile salts
Materials and methods
Chemicals
Taurocholate (2-[3α,7α,12α-trihydroxy-24-oxo-5β-cholan-24-yl) amino]ethanesulfonic acid), taurochenodeoxycholate (2-([3α,7α-dihydroxy-24-oxo-5β-cholan-24-yl]amino) ethanesulfonic acid), taurodeoxycholate (2-([3α,12α-dihydroxy-24-oxo-5β-cholan-24-yl]amino)ethanesulfonic acid), glycocholate (3α,7α,12α-trihydroxy-5β-cholan-24-oic acid N-(carboxymethyl)amide), glycodeoxycholate (3α,12α-dihydroxy-5β-cholan-24-oic acid N-(carboxymethyl)amide) and glycochenodeoxycholate (3α,7α-dihydroxy-24-oxo-5β-cholan-24-oic acid N-(carboxymethyl)amide) and γCD was all purchased from SigmaAldrich (St. Louis, USA) and used as provided. Sodium phosphate and D2O were obtained from SigmaAldrich. The water used in the experiments was obtained from a Millipore purification system.
NMR
1D 1H and 13C spectra and 2D HSQC, HMBC and ROESY spectra of 10 mM γCD mixed with 10 mM GC, GDC or GCDC in D2O, as well as 1D 1H and 13C spectra of 10 mM γCD, were recorded at 25 °C on a Bruker Avance-600 NMR spectrometer operating at 14.1 Tesla and equipped with a cryogenic cooled probe. We have previously recorded and assigned the spectra of the free BSs [21] and the spectra of the bound BSs were assigned from the recorded HSQC and HMBC spectra. 13C complexation induced shifts (CIS) were calculated by subtracting the chemical shifts of the bound BSs from the free BSs.
Molecular modeling
The free energy of the interaction between the bile salts and the γCD were calculated with a physics based molecular mechanics method using the harmonic oscillator approximation to integrate the relevant energy wells of the potential energy surfaces of the joint CD-bile salt and the free species.
The overall change in potential energy includes the anharmonic energy potentials for the non-bonding interactions and torsions, why the method takes the anharmonic effects of potential energy functions for the non-bonded interactions into consideration in the calculation of the force constants of the vibrations for each conformational energy minima. The calculations were carried out with a simplified implementation of the second-generation Mining Minima method, also denoted MS [22, 23], locally implemented in the MOE molecular mechanics platform [24], as previously described in detail [25]. The simplification in the model lead to exclusion of the hard degrees of freedom like bond stretching and angle bending and assumes harmonic energy wells. The fundament for this simplification is the previously reported insignificant entropy loss compared to the loss of the soft degrees of freedom, being the restriction imposed on the torsion angles and the loss of external rotational and translational freedom of the ligand upon binding [26]. The Boltzmann weighting of the individual chemical potentials gives a probability distribution describing the likelihood of a given complex conformation. In addition the method allows for a calculation of the total Gibbs free energy of binding when the computed chemical potential of the free cyclodextrin and bile salt are subtracted from the total chemical potential of the corresponding complex calculated over the ensemble of probability weighted complexes:
The ensembles of the free bile salts were generated by conformational analysis using the MMFF94s force field [27–33] in combination with a Generalized Born solvation model as implemented in MOE, which also included a parameterized model hydrogen bonds like the ones that can be seen for the host–guest systems evaluated in the present work. The change in the non-polar and hydrophobic solvation energy term is approximated by the change in water accessible hydrophobic surface area between the free cyclodextrin and bile salt and their mutual complex, ΔASAapolar, times a factor, b apolar, of 0.0025 kJ/mol Å2 (0.006 kcal/mol Å2) [22].
Different conformers within 12.5 kJ/mol (3 kcal/mol) of the global energy minimum were retained and subsequently docked into crystal structures of the cyclodextrin. The γCD molecule was obtained from the crystal structure with the Cambridge Structural Database code KUTKOZ. The structures were prepared for molecular docking by removal of the co-crystallized ligand molecule and manual inspection of the bond definitions and atom types. For each of the bile salt/γCD complexes 500 docking solutions were generated using MOE. Each complex was energy minimized and duplicates, defined as those having a root mean squared distance <0.05 Å, was removed. The computations of the chemical potential for the free bile salts and the bile salts in complex with the γCD were performed over the ensembles of the unique conformers of these species. Because of the complications arising from the cyclic and symmetrical structure of the γCD molecule the change in entropy for this species was ignored and only the change in conformational energy was included in the calculation of the Gibbs free energy of binding. The solvation energy correction due to the hydrophobic effect was calculated as b apolar. ΔASAapolar across the Boltzmann weighted conformation ensembles of the different species and summed with the chemical potentials to yield the total change in the calculated Gibbs free energy of binding.
Isothermal titration calorimetry
Microcalorimetric titrations were performed at 25 °C and atmospheric pressure in an aqueous 50 mM phosphate buffer solution, pH 7.0, using a MicroCal VP-ITC titration microcalorimeter (MicroCal, Northhampton, MA, USA). The isothermal titration calorimeter was calibrated electronically. The calorimetric cells was loaded with 0.9 mM solution of the bile salts and titrated with twenty-five 10 μl aliquots of 13.5 mM CD solution with 200s separation. All the solutions were degassed using a ThermoVac (MicroCal, Northhampton, MA, USA) before the titration experiment. The bile salt concentration was selected to ensure measurements below the critical micelle concentration (cmc), which for bile salts have been reported in the range of 2–10 mM [16, 18, 34–38]. This ensured that the obtained enthalpy change represented the complex formation without contributions from a concomitant dissolution of micellar aggregates. The ITC data were analysed by standard binding models and a non-linear regression routine implemented in the ORIGIN software (version 7.0). In all cases, the simplest model was used, which assumes one class of equal independent binding sites and provides the binding constant (Ks), binding enthalpy (ΔH) and the stoichiometry (n). This model accounted well for the observed enthalpograms. The standard free energy (ΔG°) and the standard entropy (ΔS°) were calculated from the standard relations to:
where R is the gas constant and T is the absolute temperature. The heat of dilution was estimated from the heat change at high CD concentrations, where practically all bile salt had been bound, and subtracted from the raw data prior to non-linear regression analysis.
Results and discussion
The inclusion of bile salts by CDs may potentially affect the oral availability. Drugs complexed to CD are dependent on displacement of the drug from the CD cavity by bile salts when a drug is coadministratered with CDs. The propensity for such displacement to occur may be evaluated by the stability constants for complexes of γCD and the relevant glyco- and tauroconjugated bile salts. This study presents a collective thermodynamic and structural description of the interaction between the six major bile salts present in the intestine of rats, dogs and humans [20] and γCD. Further, the experimental setup and the bile salts used here are the same as those in our earlier work [25], consequently it follows that the results may form the basis for a comparative analysis, which can elucidate interrelationships of chemical modification and differences between βCD and γCD and the binding of bile salt.
NMR and molecular modeling of the inclusion complexes
The interaction between bile salts and γCD have previously been reported to be a 1:1 complex [15, 17, 18], which is in accordance with the findings in the present study. In order to study the structure of these 1:1 complexes two experimental techniques are employed, ROESY NMR and Complexation Induced Shifts (CIS) based upon changes in the 13C NMR spectra. ROESY NMR is a two dimensional NMR technique, in which cross peaks of protons with intra- and inter-molecular distances of <3-4 Å can be observed [39]. CIS is another measure of where the interactions take place, which have previously been used to study the interaction between bile salts and modified or natural βCDs [40–45]. Those nuclei that take part in the complexation experience a change in environment, which can be measured as a change in their NMR chemical shifts, induced by the complexation. The nuclei that are not part of the binding site are not expected to show significant CIS. These techniques thus have the potential to determine the specific mode of interaction of the CD and guest. Cabrer et al. [15] have previously investigated γCD and bile salts with 1H-ROESY NMR where the interactions of GC, TC, GDC and TDC was studied. Their work did not identify differences caused by the bile salt conjugation. Therefore ROESY and CIS were only determined for the glycoconjugates: GC, GDC and GCDC in the present study.
CIS of all 13C nuclei in γCD and the BSs GC, GDC and GCDC are presented in Table 1. Most 13C nuclei on the BSs experience significant CIS, meaning that most parts of the BS molecules are affected by the complexation. There are important differences between the CIS induced by complexation with γCD and those previously reported for complexation with βCD [21, 25, 40]. The CIS of nuclei at the A- and B-rings and part of the C ring (nuclei 1−11 + 19) are larger when the BSs are complexed with γCD than with βCD. In contrast to this, the nuclei at the D-ring and the side chain exhibit smaller CIS when complexed with γCD compared to βCD. These observations indicate that γCD tend to encroach further onto the steroid body of the BSs than βCD, which mainly binds to the D-ring and the side chain [21, 25, 40].
A partial 1H-ROESY NMR spectra for the interaction between GCDC and γCD is presented in Fig. 2, while the two other spetra can be found in the supporting information. The conclusions derived from 13C NMR were supported by the ROESY spectra. In all spectra the strongest intermolecular crosspeaks were observed between the intracavity CD protons, H3 and H5, and the BS protons on the methyl groups P18, P19 and P21. H3 has the strongest interaction with P19 but does not seem to interact with P18 and P21 of GDC and GCDC. Only in the complex with GC an interaction between H3 and P18 is observed. In all spectra crosspeaks between H5 and P18, P19 and P21 are observed but in the complexes with GDC and GCDC the crosspeaks with P19 are stronger than in the GC complex, which on the contrary show stronger interactions between H5 and P21. These observations show that deep inclusion complexes are formed with all bile salts but GDC and GCDC are included slightly deeper than GC. In addition to these crosspeaks, all spectra show a large number of intermolecular crosspeaks between H3 and H5 and various protons on the steroid body of the BS. H5 primarily interacts with protons on the C- and D-rings and H3 also interacts with protons on the A-ring, except in the case of GC where it is not possible to identify interactions between H3 and the A-ring. H6, which is located at the primary rim of the CD, interacts with P18, P21 and P23 and supports the conclusion that the BS side chain exits the CD from the secondary rim. The suggested complex structures are similar to the complexes formed with βCD but γCD primarily resides on the steroid body whereas βCD is restricted to the sidechain, the D-ring and part of the C-ring. It is likely that the γCD complexes are less restricted and allows γCD to move back and forth on the steroid body, which leads to the interactions with several of the rings in the steroid body of the bile salts.

Partial ROESY NMR spectrum of GCDC and γCD. The region of mostly bile salt chemical shift is depicted at the x-axis and the region of CD chemical shift at the y-axis. BS protons are denoted with “P” and Cd protons with “H”
The modelling showed the interaction included the C ring of the steroid skeleton of the bile salt completely and the B and D ring partly, see Fig. 3. This observation is in accordance with the binding suggested by Cabrer et al. [15]. However, for TC/GC and TDC/GDC the modelling also suggested a different orientation with the conjugation of the bile salt out of the CD from both the primary side, see Fig. 4. This was neither supported by the chemical shifts observed in the 13C NMR nor the 1H-ROESY NMR. As there is a lack of experimental data supporting the modelling on this point, the conclusions should be kept with this precaution on mind. However, the possible orientations predicted by the modelling could be interpretated as an indication of a less restricted interaction than the similar interaction with βCD, in that bile salt can be oriented in both directions in the CD cavity.

Most stable conformation for complexes with γCD and a GCDC and b GC. γCD are presented as the stick model and the bile salts as the space filling model

Inclusion of GC with γCD in the two suggested orientations. a The most stable orientation with exit of the bile salt conjugation from the secondary ring; and b the less stable orientation with exit of the bile salt conjugation from the primary ring. CD are presented as the stick model and the bile salts as the space filling model. Independent of orientation the molecular modeling suggest that γCDs make complexes with bile salt where the bile salt D- and B-rings are almost entirely included in the cyclodextrin interior, whereas the C-ring is entirely included
Stability constants
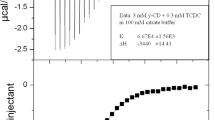
ITC was used to study the interaction of the six bile salts and γCD at 25 °C. A typical thermogram following an ITC titration is presented in Fig. 5. The stability constants obtained through the ITC experiments for the six different complexes are presented in Table 2 together with the available literature data measure by the same method. The affinity for TDC and TCDC was slightly lower than its glycol-conjugated counterparts, GDC and GCDC, whereas no difference was seen between TC and GC.

Calorimetric titration of TC with γCD in 50 mM phosphate buffer (pH 7.0) at 25 °C. Raw data for sequential 10 μl injections of γCD solution (10 mM) into the bile salt solution (1 mM). Heats of reaction as obtained from the integration of the calorimetric traces
The stability constant for the binding of the two trihydroxy bile salts TC and GC to γCD was just over 5,000 M-1, see Table 2. The stability constant for TDC and GDC was significantly higher and even higher for TCDC/GCDC. The strength of the stability constant for TCDC/GCDC is at a size, where it could be considered to isolate crystals of the complex for X-ray investigations, which could be a topic for future studies. The obtained stability constants was in range with previously published results generated by different techniques [15–17, 19] verifying the results obtained in the present work. The binding strengths of the different kinds of bile salts show that the number and positions of hydroxyl groups on the steroid body of the bile salts has a strong influence on the complex stabilities, as previously discussed by Holm and coworkers [17]. Both the NMR studies and the modeling demonstrated the interaction between the bile salts and γCD to occur with full inclusion of the bile salts C-ring. TCDC/GCDC lack a hydroxyl group on C12, making this interaction easier and hence stronger than the two other bile salt classes, which has a hydroxyl group in this position. Comparing the chemical structure of TC/GC with TDC/GDC reveals a hydroxyl group less on the B-ring of the latter. As both NMR and modeling demonstrated interactions with the B ring, a hydroxyl group there appears to obstruct the interaction.
The ranking and relative size of the stability constants is similar to what is obtained for the binding of the same bile salts to βCD [25, 46]. This is despite that Holm et al. [25] demonstrated complexation on the bile salts D-ring when interacting with βCD by ROESY NMR and molecular modeling, whereas the interaction with γCD occurs on the D-, C- and B-ring. GDC/TDC has a higher stability constant to γCD than to βCD, probably do to a different inclusion into the larger cavity of the γCD, but still the binding strengths of the two different CDs must be considered relatively similar, which despite the different binding positions, could indicate common driving forces in the interaction with bile salts, e.g. similar hydration and electrostatic effects [47].
Enthalpy and entropy changes
The interaction was exothermic in all cases (Table 2), and the size of the enthalpy contribution was in good agreement with the previous reported data for TDC [18]. As discussed in the previous section the stability constants were highly affected by the position of the hydroxyl groups on the bile salts. This may be further elucidated by the enthalpy and entropy data as these are generally more sensitive to details in the mode of interaction than the stability constants, due to the characteristic enthalpy–entropy compensation, which tends to hide effects on the level of the free energy changes (or binding constants). Thus, the entropic contribution to the complex formation is about twice as large for the TCDC/GCDC and TDC/GDC compared to TC/GC (Table 2).The two former are dihydroxy bile salts while the latter is a trihydroxy bile salt. Also the enthalpic contribution was different. One way to analyse these differences is through a so-called compensation plot, which shows the enthalpy change as a function of the entropy change. It appears from Fig. 6a that the four weakly-binding bile salts (the cholates and deoxycholates) fall on a line (dashed line, R2 = 0.992). A linear correlation of ΔS and ΔH in a homologous series is normally considered as an indication that the members of the series share a single source of additivity [48]. In the current context, this suggests that TC, GC, TDC and GDC bind to γCD with a common mechanism, but with different contributions to ΔS and ΔH dictated by the detailed chemical structure of the ligand. Conversely, Fig. 5 suggests that TCDC/GCDC show a qualitatively different mode of interaction, which was not strongly supported by the structural analysis nor the 1H-ROESY NMR, but could be a reflection of the lacking hydroxyl group on C12.

Compensation plot of the individual data points showing the enthalpy change as a function of the entropy change for a) the investigated γCD complexes and b) the γCD and βCD complexes from Holm et al. [25]. The bile salts are indicated by labels in the plot. The dashed line is a linear plot to the results (see main text for details)
Comparing the present data for γCD with the results for βCD and the same bile salts [25] shows differences in the thermodynamics. The interaction for βCD is mostly enthalpy driven with a relative large entropic penalty. Conversely for γCD we observe a favourable entropic contribution to the complexation. For both β- and γCD, inclusion of the bile salt expels water from the cavity, which is thought to contribute positively to entropy [49], under the assumption that water is more ordered in the cavity than in the bulk. This would cause a larger increase in entropy in the case of γCD with a larger cavity. The higher enthalpic contribution for βCD could also originate from the smaller cavity size of βCD, which will in general give shorter interaction distances and hence lower energy. However, this narrower fit will also lower the entropy, by locking the complex tighter contributing to the unfavourable entropies of the βCD complexes. Similar comparison to the interaction between bile salts and αCD is not possible, as this complexation is very weak [15, 18, 50, 51]. In Fig. 6b the compensation plot includes data for both β- and γCDs interacting with bile salts. As mentioned above, placement on the same line is an indication of a single source of additivity [48]. It has been suggested that the slope and the intercept of the enthalpy–entropy compensation plot can be related to the degree of conformational change and the extent of desolvation induced upon complexation, respectively [52–55]. Figure 6b suggests a relative large intercept with the x-axis for both classes of CDs and bile salts. This large intercept indicates an extensive desolvation effect in the interaction between the investigated species, i.e. a common driving force for the two CDs in the interaction.
Differences in the buried polar and hydrophobic surface areas are also reflected in the binding affinities. The theoretically calculated ASA for the bile salts and the bile salt-CD complex are presented in Table 3. Reduction of ASA for the hydrophobic parts (ASAapolar) of the molecules is perceived as thermodynamically favorable, i.e. the larger the reduction the stronger the (hydrophobic) attraction. In contrast to this, reduction of ASA for the polar components is thermodynamically unfavorable, i.e. the smaller reduction the better. This correlation between stability and changes in polar and nonpolar surface area is indeed found here. Thus, the most stable complexes (TCDC and GCDC, Table 2) show the largest ΔASAapolar and the smallest ΔASApolar (Table 3). The opposite is found for the most weakly bound bile salts (TC and GC). The differences observed in the changes in the two ASA values for the interaction between the different bile salts and γCD therefore indicate that the variations in ΔS may, at least partly, be ascribed to differences in the hydration effects. The difference in the entropy changes could also be a reflection of the changes in water accessible surface area (ASA) when the complex forms. When the water is transferred from the hydration shell of a non-polar moiety to the bulk a strong positive contribution to ΔS is observed. Therefore, the higher values of ΔS for TCDC, GCDC, TDC and GDC could also reflect a more pronounced hydrophobic interaction (dehydration of more non-polar ASA) for these complexes. A linear fit of TΔS plotted against ΔASAapolar, see Fig. 7, has a slope of −241 ± 32 J/mol/Å2. Costas et al. [55] have calculated that each Å2 of a non-polar molecule that is hydrated contributes with −231 J/mol to TΔS. The similarity of these two values thus suggests that differences in ΔASAapolar between the complexes are the main contributor to the differences in ΔS.

Correlation between TΔS° and non-polar dehydrated surface area of the complexes, the black line shows the best fit to the data
When the changes in ASA for γCD from the present study is compared to similar calculations for βCD [25] a smaller and higher change is observed for ASApolar and ASAhyd, respectively. This in general indicates more favorable entropy for the interaction between bile salts and γCD when compared to the interaction with βCD, which is in accordance with the experimental results reported for the two different systems. This systematic difference between the two classes of CDs combined with the NMR and the ITC data clearly suggest that the insertion of the B, C and D ring of the bile salt molecule leads to a more effective dehydration of the apolar part of the bile salt molecule and therefore a more favorable ΔS when interacting with γCD
Conclusions
In conclusion, this study has presented 1:1 stability constants and thermodynamic parameters for the binding of six biologically relevant glyco- and tauro-conjugated bile salts to the natural γCD. 1H-ROESY NMR experiments, supported by molecular modeling, demonstrated the interaction to include the C-ring of the bile salt completely into the complex and the B- and D-ring partly irrespectively of the bile salt type.
ITC was successfully applied for the investigation of the bile salt–CD interactions. All of the investigated bile salts had a significant affinity for γCD. The complexation was found to be driven by both enthalpy and entropy for all the bile salts, which was in contrast to reported data on βCD. This is attributed to the larger non-polar surface area dehydrated upon complexation with the larger γCD. The presence of hydroxyl groups at position C12 strongly affected the affinity of the bile salts for the CDs and lead to a lower stability constant.
References
Szejtli, J.: Cyclodextrin Technology. Kluwer, Dordrecht (1988)
Loftsson, T., Brewster, M.E., Masson, M.: Role of cyclodextrins in improving oral drug delivery. Am. J. Drug Deliv. 2, 175–261 (2004)
Carrier, R.L., Miller, L.A., Ahmed, I.: The utility of cyclodextrins for enhancing oral bioavailability. J. Control. Release. 123, 78–99 (2007)
Davis, M.E., Brewster, M.E.: Cyclodextrin-based pharmaceutics: past, present and future. Nat. Rev. Drug. Discov. 3, 1023–1035 (2004)
Brewster, M.E., Loftsson, T.: Cyclodextrins as pharmaceutical solubilizers. Adv. Drug. Deliv. Rev. 59, 645–666 (2007)
Rajewski, R.A., Stella, V.J.: Pharmaceutical applications of cyclodextrins. 2. in vivo drug delivery. J. Pharm. Sci. 85, 1142–1169 (1996)
Thompson, D.O.: Cyclodextrins-enabling excipients: their present and future use in pharmaceuticals. Crit. Rev. Ther. Drug. Carrier. Syst. 14, 1–104 (1997)
Uekama, K.: Design and evaluation of cyclodextrin-based drug formulation. Chem. Pharm. Bull. 52, 900–915 (2004)
Westerberg, G., Wiklund, L.: β-cyclodextrin reduces bioavailability of orally administered [3H]benzo[a]pyrene in the rat. J. Pharm. Sci. 94, 114–119 (2005)
Uekama, K., Hirayama, F., Irie, T.: Cyclodextrin drug carrier systems. Chem. Rev. 98, 2045–2076 (1998)
Ono, N., Hirayama, F., Arima, H., Uekama, K., Rytting, J.H.: Model analysis for oral absorption of a drug/cyclodextrin complex involving competitive inclusion complexes. J. Incl. Phenom. Macrocycl. Chem. 44, 06–93 (2003)
Marshall, J.J.M.: Kinetic difference between hydrolyses of γ-cyclodextrin by human salivary and pancreatic α-amylase. BBA 661, 142–147 (1981)
Kondo, H., Nakatani, H., Hiromi, K.: In vitro action of human and porcine α-amylase on cyclo-malto-oligosaccharides. Carbohydr. Res. 204, 207–213 (1990)
Lipindski, C.A.: Drug-like properties and the causes of poorly solubility and poor permeability. J. Pharmacol. Toxicol. Methods 44, 235–249 (2000)
Cabrer, P.R., Alvarez-Parrilla, E., Al-Soufi, W., Meijide, F., Núñez, E.R., Tato, J.V.: Complexation of bile salts by natural cyclodextrins. Supramol. Chem. 15, 33–43 (2003)
Tan, Z.J., Zhu, X.X., Brown, G.R.: Formation of inclusion complexes of cyclodextrins with bile salt anions as determined by NMR titration studies. Langmuir 10, 1034–1039 (1994)
Holm, R., Hartvig, R.A., Nicolajsen, H.V., Westh, P., Østergaard, J.: Characterization of the complexation of tauro- and glyco-conjugated bile salts with γ-cyclodextrin and 2-hydroxypropyl-γ-cyclodextrin using affinity capillary electrophoresis. J. Incl. Phenom. Macrocycl. Chem. 61, 161–169 (2008)
Cooper, A., Nutley, M.A., Camilleri, P.: Microcalometry of chiral surfactant–cyclodextrin interactions. Anal. Chem. 70, 5024–5028 (1998)
Abadie, C., Hug, M., Kübli, C., Gains, N.: Effect of cyclodextrins and undigested starch on the loss of chenodeoxycholate in the feces. Biochem. J. 229, 725–730 (1994)
Alvaro, D., Cantatore, A., Attili, A.F., Gianni Corrandini, S., De Luca, C., Minervini, G., Di Blase, A., Angelico, M.: Relationships between bile salts hydrophilicity and phosphorlipid composition in bile of various animal species. Comp. Biochem. Physiol. 83B, 551–554 (1986)
Schonbeck, C., Westh, P., Madsen, J.C., Larsen, K.L., Stade, L.W., Holm, R.: Methylated beta-cyclodextrins: influence of degree and pattern of substitution on the thermodynamics of complexation with tauro- and glyco-conjugated bile salts. Langmuir 27, 5832–5841 (2011)
Chen, W., Chang, C.-E., Gilson, M.K.: Calculation of cyclodextrin binding affinities: energy, entropy, and implication for drug design. Biophys. J. 87, 3035–3049 (2004)
Chang, C.E., Gilson, M.K.: Free energy, entropy, and induced fit in host-guest recognition: calculations with the second-generation mining minima algorithm. J. Am. Chem. Soc. 126, 13156–13164 (2004)
Molecular Operating Environment (MOE), Version 2006.08 [Computer Program]. Montreal (2002)
Holm, R., Shi, W., Hartvig, R.A., Askjær, S., Madsen, J.C., Westh, P.: Thermodynamics and structure of inclusion compounds of tauro- and glyco-conjugated bile salts and β-cyclodextrins. Phys. Chem. Chem. Phys. 11, 5070–5078 (2009)
Chang, C.E., Chen, W., Gilson, M.K.: Ligand configurational entropy and protein binding. Proc. Natl Acad. Sci. USA 104, 1534–1539 (2007)
Halgren, T.A.: Merck molecular force field. 1. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 17, 490–519 (1996)
Halgren, T.A.: Merck molecular force field. 2. MMFF94 van der waals and electrostatic parameters for intermolecular interactions. J. Comput. Chem. 17, 520–552 (1996)
Halgren, T.A.: Merck molecular force field. 3. Molecular geometries and vibrational frequencies for MMFF94. J. Comput. Chem. 17, 553–586 (1996)
Halgren, T.A., Nachbar, R.B.: Merck molecular force field. 4. Conformational energies and geometries for MMFF94. J. Comput. Chem. 17, 587–615 (1996)
Halgren, T.A.: Merck molecular force field. 5. Extension of MMFF94 using experimental data, additional computational data, and empirical rules. J. Comput. Chem. 17, 616–641 (1996)
Halgren, T.A.: MMFF VI. MMFF94s option for energy minimization studies. J. Comput. Chem. 20, 720–729 (1999)
Halgren, T.A.: MMFF VII. Characterization of MMFF94, MMFF94s, and other widely available force fields for conformational energies and for intermolecular-interaction energies and geometries. J. Comput. Chem. 20, 730–748 (1999)
Hofmann, A.F., Roda, A.: Physicochemical properties of bile acids and their relationship to biological properties: an overview of the problem. J. Lipid Res. 25, 1477–1489 (1984)
Roda, A., Hofmann, A.F., Mysels, K.J.: The influence of bile salt structure on self-association in aqueous solutions. J. Biol. Chem. 258, 6362–6370 (1983)
Jana, P.K., Moulik, S.P.: Interaction of bile salts with hexadecyltrimethylammonium bromide and sodium dodecyl sulfate. J. Phys. Chem. 95, 9525–9532 (1991)
Funasaki, N., Ueshiba, R., Hada, S., Neya, S.: Stepwise self-association of sodium taurocholate and taurodeoxycholate as revealed by chromatography. J. Phys. Chem. 98, 11541–11548 (1994)
Garidel, P., Hildebrand, A., Neubert, R., Blume, A.: Thermodynamic characterization of bile salt aggregation as a function of temperature and ionic strength using isothermal titration calorimetry. Langmuir 16, 5267–5275 (2000)
Schneider, H.-J., Hacket, F., Rüdiger, V.: NMR studies of cyclodextrins and cyclodextrin complexes. Chem. Rev. 98, 1755–1785 (1998)
Holm, R., Madsen, J.C., Shi, W., Larsen, K.L., Städe, L.W., Westh, P.: Thermodynamics of complexation of tauro- and glyco-conjugated bile salts with two modified β-cyclodextrins. J. Incl. Phenom. Macrocycl. Chem. 69, 201–211 (2011)
Mucci, A., Schenetti, L., Vandelli, M.A., Ruozi, B., Salvioli, G., Forni, F.: Comparison between Roesy and 13C NMR complexation shifts in deriving the geometry of inclusion compounds: a study on the interaction between hyodeoxycholic acid and 2-hydroxypropyl-β-cyclodextrin. Supramol. Chem. 12, 427–433 (2001)
Mucci, A., Vandelli, M.A., Salvioli, G., Malmusi, L., Forni, F., Schenetti, L.: Complexation of bile salts with 2-hydroxypropyl-β-cyclodextrin: a 13C NMR study. Supramol. Chem. 7, 125–127 (1996)
Mucci, A., Schenetti, L., Salvioli, G., Ventura, P., Vandelli, M.A., Forni, F.: The interaction of biliar acids with 2-hydroxypropyl-β-cyclodextrin in solution and in the solid state. J. Incl. Phenom. Macrocycl. Chem. 26, 233–241 (1996)
Vandelli, M.A., Salvioli, G., Mucci, A., Panini, R., Malmusi, L., Forni, F.: 2-Hydroxypropyl-β-cyclodextrin complexation with ursodeoxycholic acid. Int. J. Pharm. 118, 77–83 (1995)
Vandelli, M.A., Ruozi, B., Forni, F.: A solution and solid state study on 2-hydroxypropyl-β-cyclodextrin complexation with hyodeoxycholic acid. J. Incl. Phenom. Macrocycl. Chem. 37, 237–251 (2000)
Holm, R., Nicolajsen, H.V., Hartvig, R.A., Westh, P., Østergaard, J.: Complexation of tauro- and glyco-conjugated bile salts with three neutral β-cyclodextrins studied by affinity capillary electrophoresis. Electrophoresis 28, 3745–3752 (2007)
Liu, L., Guo, Q.-X.: The driving force in the inclusion complexation of cyclodextrins. J. Incl. Phenom. Macrocycl. Chem. 42, 1–14 (2002)
Lumry, R.: On the interpretation of data from isothermal processes. Methods. Enzmol. 259, 628–720 (1995)
Jelesarov, I., Bosshard, H.R.: Isothermal titration calorimetry and differential scanning calorimetry as complementary tools to investigate the energetics of biomolecular recognition. J. Mol. Recognit. 12, 3–18 (1999)
Holm, R., Schönbeck, C., Askjær, S., Jensen, H., Westh, P., Østergaard, J.: Complexation of tauro- and glyco-conjugated bile salts with α-cyclodextrin and hydroxypropyl-α-cyclodextrin studied by affinity capillary electrophoresis and molecular modelling. J. Sep. Sci. 34, 3221–3230 (2011)
Inoue, Y., Hakushi, T., Liu, Y., Tong, L.-H., Shen, B.-J., Jin, D.-S.: Thermodynamics of molecular recognition by cyclodextrins. 1. Calorimetric titration of inclusion complexation of naphthalenesulfonates with α, β and γ-cyclodextrins: enthalpy–entropy compensation. J. Am. Chem. Soc. 115, 475–481 (1993)
Inoue, Y., Lin, Y., Tong, L.-H., Shen, B.-J., Jin, D.-S.: Thermodynamics of molecular recognition by cyclodextrins. 2. Calorimetric titration of inclusion complexation with modified β-cyclodextrins. Enthalpy–entropy compensation in host-guest complexation: from ionophone to cyclodextrin and cyclophane. J. Am. Chem. Soc. 115, 10637–10644 (1993)
Rekharsky, M.V., Inoue, Y.: Complexation thermodynamics of cyclodextrins. Chem. Rev. 98, 1875–1917 (1998)
Rekharsky, M.V., Mayhew, M.P., Goldberg, R.N., Ross, P.D., Yamashoji, Y., Inoue, Y.: Thermodynamic and nuclear magnetic resonance study of the reactions of alpha- and beta-cyclodextrin with acids, aliphatic amines, and cyclic alcohols. J. Phys. Chem. B 101, 87–100 (1997)
Costas, M., Kronberg, B., Silveston, R.: General thermodynamic analysis of the dissolution of nonpolar molecules into water–origin of hydrophobicity. J. Chem. Soc. Faraday Trans. 90, 1513–1522 (1994)
Acknowledgments
Erik L. Rasmussen is acknowledged for skilfulled technical help with the ITC measurements.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
10847_2012_165_MOESM1_ESM.pdf
The online version of this article contains supplementary material, which is available to authorized users. (PDF 169 kb)
Rights and permissions
About this article
Cite this article
Holm, R., Schönbeck, C., Askjær, S. et al. Thermodynamics of the interaction of γ-cyclodextrin and tauro- and glyco-conjugated bile salts. J Incl Phenom Macrocycl Chem 75, 223–233 (2013). https://doi.org/10.1007/s10847-012-0165-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10847-012-0165-1