
Abstract
The synthesis of salicylic acid derivatives of 1,4,7,10-tetraazacyclododecane has been described. The complexing properties of these compounds towards metal cations were investigated by absorption and emission spectroscopy, and 1H NMR. The X-ray structure of L-2 nitrate is reported.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
1,4,7,10-Tetraazacyclododecane derivatives have demonstrated a broad spectrum of uses, including chemistry, pharmaceuticals, medicine, and environmental protection [1–4]. Conceptually the simplest and most convergent method of preparation mono and disubstituted ligands is direct alkylation or acylation of cyclen [5–8]. Although one-pot procedure is simple, the three-step procedure in which protected 1,4,7,10-tetraazacyclododecane is prepared first, followed by alkylation or acylation reaction followed by deprotection, has generally been found to result in higher yields [9–11].
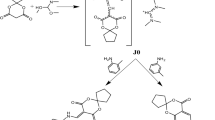
We are particularly interested in direct synthesis of hydroxy acid derivatives of 1,4,7,10-tetraazacyclododecane. Hydroxy acids and their derivatives are known as efficient chelating reagents for metal cations. For example, salicylic acid forms complexes with Al+3, Fe+3, Cu+2, Co+2, Mg+2 and Ca+2 [12]. Introduction of hydroxy acid moieties to macrocycle should efficiently modify complexing ability of entire molecule. So far three 1,4,7,10-tetraazacyclododecane derivatives containing salicylic acid residue were synthesized (Fig. 1) [13, 14]. Compounds 1, 2 and 3 have light-harvesting moieties but only ligand 1 exhibits moderate fluorescence. UV–Vis Spectroscopic studies show that compound 1 forms complexes only with Co2+ ions while both compounds 2 and 3 are able to complex Mg+2, Ca+2, and Sr+2 cations. The obtained results suggest that the structure of substituent (salicylic acid derivative) is crucial for selective cation recognition. In this paper we present synthesis and spectroscopic studies of new salicylic acid derivatives of cyclen.

Synthesized salicylic acid derivatives of 1,4,7,10-tetraazacyclododecane
Experimental
General
1H NMR and 13C NMR spectra were recorded at 500 MHz and 125 MHz on Varian instrument, respectively. Mass spectra were recorded using a VG Platform II electrospray mass spectrometer with methanol as a carrier solvent. Elemental analysis was performed on Carlo Erba CHNS-O EA 1108 apparatus. UV spectra and fluorescence emission spectra were recorded on a Perkin Elmer Lambda 40P spectrophotometer and a Perkin Elmer LS-50B fluorescence spectrophotometer, respectively.
Thin layer chromatography (TLC) analyses were carried out on Alufolien covered with silica gel 60-F-254 (0.2 mm thickness) while silica gel 60 (70–230 mesh) was used for column chromatography.
All reagents and solvents were of the highest commercial quality and were used without further purification. Zn(ClO4)2 · 6H2O, Cu(ClO4)2 · 6H2O, Co(ClO4)2 · 6H2O, Ni(ClO4)2 · 6H2O, Cd(ClO4)2 · 6H2O, Al(ClO4)3 · 9H2O and Al(NO3)3 · 9H2O were purchased from Aldrich. All salts were dried under vacuum at 60 °C before use. Acetonitrile was dried over calcium hydride before use. Water refers to high purity water with conductivity of ≤0.03 μS cm−1, obtained from the HYDROLAB purification system. 1,4,7,10-Tetraazacyclododecane (Strem Co., France), salicylic acid and 4-nitrophenol (Aldrich) are commercially available and were used as received.
Potentiometric titrations were performed in water at 25 °C using an OP-205 Radelkis pH-meter linked to a personal computer. All the test solutions (50 mL) were kept under argon (>99.999% purity) atmosphere. The potentiometric pH titrations were carried out with I = 0.1 (NaNO3) at 25.0 ± 0.1 °C, and at least two independent titrations were performed. Protonation constants were determined by means of the STOICHIO program [15].
UV–Vis spectra and fluorescence emission spectra were recorded at 25.0 ± 0.1 °C on a Perkin Elmer Lambda 40P spectrophotometer and a Perkin Elmer LS-50B fluorescence spectrophotometer, respectively. For fluorescence titration, a sample solution was excited at 310 nm or 294 nm if L-1 was studied, and 288 nm or 283 nm if L-2 was used. The obtained data from UV titrations were analyzed for apparent complexation constants K using the STOICHIO program [15].
1-N-Salicyloyl-1,4,7,10-tetraazacyclododecane (L-1) and 1,7-bis-N,N′-salicyloyl-1,4,7,10-tetraazacyclododecane (L-2)
A stoichiometric amount of 4-nitrophenyl active ester [16] (5 mmol, 1.3 g) was added to a solution of cyclen (5 mmol, 0.9 g) in dry THF (15 mL). The reaction mixture was stirred for 10–12 h at room temperature. The solvent was evaporated under vacuum, and the oily residue was purified by gradient column chromatography, using solvent system CHCl3/MeOH/NH3. Fraction, which was eluted with 40:1:0.1 (CHCl3:MeOH:NH3) mixture was collected giving L-2 (0.3 g, 20%, R f = 0.25) as a colorless oil after solvent evaporation. Further increasing of solvent polarity up to 20:4:1 (CHCl3:MeOH:NH3) allowed separation of L-1 (0.9 g, 60%, R f = 0.19), as a colorless oil.
L-1: 1H NMR (d-DMSO): 2.48–2.49 (m, 2H), 2.54–2.55 (m, 2H), 2.63–2.64 (m, 2H), 2.69–2.71 (m, 2H), 2.79–2.82 (m, 4H), 3.47–3.48 (m, 4H), 5.20–5.40 (br, 4H), 6.45 (t, J = 7.3 Hz, 1H), 6.55 (d, J = 8.3 Hz, 1H), 6.97 (dd, J = 1.5 and 7.8 Hz, 1H,), 7.02 (dt, J = 1.5 and 7.3 Hz, 1H); 13C NMR: 44.34, 45.86, 46.74, 47.58, 47.70, 48.32, 49.20, 51.82, 114.97, 119.70, 126.02, 129.76, 130.81, 159.57, 172.96; HRMS [EI, (M+)] calculated for C15H24N4O2 292.18993; found 292.19035; Anal. Calcd. for: C15H24N4O2: C, 61.62; H, 8.27; N, 19.16. Found: C, 61.56; H, 8.30; N, 19.13.
L-2: 1H NMR (d-DMSO): 2.66–2.67 (m, 5H), 2.89–2.93 (m, 5H), 3.34–3.44 (m, 8H), 6.66 (t, J = 7.3 Hz, 2H), 6.70–6.72 (m, 2H), 7.05 (d, J = 7.3 Hz, 2H), 7.11–7.12 (m, 2H) 8.01 (s, 2H); 13C NMR: 45.26, 46.97, 48.07, 48.68, 50.01, 117.22, 118.22, 125.91, 128.51, 130.26, 156.24, 171.60; HRMS [EI, (M+)] calculated for C22H28N4O4 412.21106; found 412.21196; Anal. Calcd. for: C22H28N4O4: C, 64.06; H, 6.84; N, 13.58. Found: C, 64.01; H, 6.86; N, 13.56.
[L-2] · 2 (HNO3) · CH3CN preparations
1,7-Bis-N,N′-salicyloyl-1,4,7,10-tetraazacyclododecane (0.1 mmol, 41 mg) was dissolved in acetonitrile (10 mL) and Al(NO3)3 · 9H2O was added. The mixture was refluxed 2 h then left at the room temperature for several days. Single crystals were collected, washed with acetonitrile and dried.
Anal. Calcd. for: C22H28N4O4·2(HNO3)·CH3CN: C, 49.74; H, 5.74; N, 16.92. Found: C, 49.70; H, 5.80; N, 16.89. M.p. 160 °C (decomposition).
X-ray structure description
1,7-Bis-N,N′-salicyloyl-1,4,7,10-tetraazacyclododecane nitrate single crystal was grown as result of alumina nitrate hydrolysis in acetonitile during complex preparation. For X-ray studies a transparent crystal of needle shape was selected. X-ray measurements were carried out on a KUMA KM4 four-axis diffractometer equipped with a Sapphire-2 CCD detector (Oxford Diffraction) at working temperature 120 K (Oxford Cryosystem).
Data collection: CrysAlis CCD (Oxford Diffraction [17], 2005); cell refinement: CrysAlis RED (Oxford Diffraction, 2005); data reduction: CrysAlis RED; program(s) used to solve structure: SHELXS97 (Sheldrick [18], 1997); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: ORTEP-3 for Windows [19] (Farrugia, 1997); software used to prepare material for publication [20]: WinGX (Farrugia, 1999). All H atoms were positioned geometrically (C–H = 0.95–0.99 Å) and refined as riding with their Uiso values constrained to be x times Ueq of their pivot atom (x = 1.2 for aromatic C–H, 1.3 for methylene C–H and 1.5 for methyl C–H). The N–H and O–H bonds were constrained to 0.92 Å. The highest electron density peak (0.24) is located 0.86 Å from atom C1.
Crystallographic data for the structure reported in this paper has been deposited with the Cambridge Crystallographic Data Centre as Supplementary Publication No CCDC 634437.
Copies of the data can be obtained free of charge from the CCDC (12 Union Road, Cambridge CB2 1EZ, UK; Tel.:+44-1223-336-408; fax:+44-1223-336-003; e-mail: deposit@ccdc.cam.ac.uk; http://www.ccdc.cam.ac.uk).
Results and discussion
The reaction of salicylic acid chloride with 1,4,7,10-tetraazacyclododecane afforded a mixture of products. The yield of the monosubstituted product after column separation was only 10%. The use of 1,4,7-tri-tert-butoxycarbonyl-tetraazacyclododecane allowed to obtain the product after deprotection and column purification with the yield of 15%. In this situation another method was attempted. Among available acylation reagents of salicylic acid, active esters were chosen. Thus, 4-nitrophenyl active ester of salicylic acid was prepared in reaction of salicylic acid and 4-nitrophenol using POCl3 as condensing agent in toluene solution [16]. In the next step, the active ester was used in the substitution reaction (Scheme 1).

Direct acylation of cyclen with 4-nitrophenyl salicylate
Cyclen (1 mmol) and the active ester (1 mmol) were dissolved in dry THF. The solution was left for 10–12 h at room temperature. The progress of acylation was controlled by TLC. In the reaction only mono- and disubstituted products were formed, although the reagents were used in molar ratio 1:1 and high dilution condition was not applied. The yield of monosubstituted product (L-1) was 60%. It is worth to note that changes of reaction conditions such as elevated temperature 40 °C and 1:2 molar ratio (cyclen: active ester) allowed to obtain diamide as the major product (L-2) with 60% yield.
L-2 forms crystals at presence of nitrate anions in acetonitrile solution. We obtained single crystals of L-2 nitrate as results of hydrolysis reaction of Al(NO3)3 during complex preparation. X-ray study reveals that in the crystal structure of L-2 the asymmetric unit contains half of the cyclen molecule sitting at the symmetry center (Wyckoff position [21] f), one nitrate anion and one molecule of acetonitrile in general positions. Elemental cell is composed of one macrocyclic dication, two \( {\text{NO}}^{ - }_{{\text{3}}} \) ions and two molecules of CH3CN. The macrocyclic molecule carries double positive charge due to protonation of both N2 and N2′ atoms. Figure 2 shows described above structure and extra nitrate anion for better illustration of hydrogen bonds.

ORTEP plot of L-2, showing the atom labeling scheme. Displacement ellipsoids are drawn at 50% probability level. Symmetry transformations used to generate equivalent atoms: primed: −x + 1, −y, −z + 1 doubly primed: −x + 2, −y, −z + 1. One more \( {\text{NO}}^{ - }_{{\text{3}}} \) anion (equivalent) is shown to reveal hydrogen bonding connectivity
Both N–H are involved in hydrogen bonding. One is intramolecular: H2a uses O2 phenolic oxygen as the acceptor, whereas H2b forms intermolecular bond with oxygen atom O4′ from the nitrate anion. Hydrogen atom (H2c) from the phenolic O–H group forms a different intermolecular bond to another oxygen atom O3″ of the anion (Table 1). The solvent (CH3CN) is not involved into hydrogen bonding and simply fills space in cavities of the crystal structure.
The protonation constants (Kn) of L-1 and L-2 were determined by potentiometric and spectrophotometric pH titrations of the acidic solutions of L-1 (C L-1 = 4.72 × 10−4 M, CH+ = 1.17 × 10−3 M CH3SO3H) and L-2 (C L-2 = 3.03 × 10−4 M, CH+ = 8.54 × 10−4 M CH3SO3H) against 1.83 × 10−2 NaOH (L-1) and 8.94 × 10−3 M NaOH (L-2) with I = 0.1 (NaNO3) at 25 °C. The titration data were analyzed for the acid-base equilibrium 1 and 2, where a H+ is the activity of H+.
The protonation constants K n were calculated and were collected in Table 2 as pKa.
Figure 3 showed pH dependent UV titrations of ligands L-1 and L-2 (λ = 300 nm). The increase of absorbance was observed in both cases, but the shape of the curves was different. If compound L-1 was used, absorbance increase was observed in pH range 3.5–7 while the absorbance value of L-2 was almost constant. In the pH range 7–11 the increase of absorbance was observed for both ligands, but the absorption intensity of L-2 increased about 35% compared to L-1 of pH 10.

(a) pH Depended UV-pH for L-1 (C L-1 = 4.72·10−4 M, CH+ = 1.169 × 10−3 M, CNaOH = 8.943 × 10−3 M) at 25 °C with I = 0.1 (NaNO3); (b) UV-pH profile for L-2 (C L-2 = 3.03 × 10−4 M, CH+ = 8.54 × 10−4 M, CNaOH = 1.083 × 10−2 M) at 25 °C with I = 0.1 (NaNO3). The experimental points are open squares and the fitting curve for the model is a line
Complexing properties of L-1 and L-2 with metal cations were studied first in acetonitrile solution. In case of Zn2+ ions several equilibriums of very strong complexes were found for both ligands and the calculation of complexation constants was impossible. To simplify the system, acetonitrile was replaced by water. In aqueous solution L-1 (absorption maximum at 300 nm, ε = 1860 [M−1 · cm−1]) formed also several complexes with Zn2+cations. The spectroscopic UV–Vis titration is shown in Fig. 4a. The increase of the absorbance band at λ = 281 nm and decrease of the band at λ = 300 was observed. The course of titration of L-2 (absorption maximum at 300 nm, ε = 3650 [M−1 · cm−1]) was comparable to L-1 but, in the contrast, L-2 formed only one ionic complex [Zn(L-1)2]2+ for which log β value was estimated as 8.44 ± 0.09 (Fig. 4b).

(a) Absorption spectra recorded in water solution containing ligand L-1 (4.24 × 10−4 M) with Zn(ClO4)2 (0–2.27 × 10−3 M). (b) Absorption spectra recorded in water solution containing ligand L-2 (1.95 × 10−4 M) with zinc perchlorate (0–1.24 × 10−3 M)
Compound L-2 formed also complexes with Cd2+ and Al3+ cations (Fig. 5a, b). The stoichiometry of complex with Cd2+ was 1:1 and the value of log K was estimated to be 4.34 ± 0.05. In case of spectroscopic titration of L-2 with Al(ClO4)3 the formation of new band at λ = 340 nm was observed and two types of equilibrium in methanol solution was found. The stoichiometry of complexes were 1:1 and 1:2 (Al3+: L-2), but the main form was the second one.

(a) Absorption spectra recorded in water solution containing ligand L-2 (2.02 × 10−4 M) with Cd(ClO4)2 (0–1.43 × 10−3 M). (b) Absorption spectra recorded in methanol solution containing ligand L-2 (c = 1.68 × 10−4 M) with Al(ClO4)3 (c = 0–8.32 × 10−4 M)
Complex formation has also been studied by 1H NMR. The main problems in these experiments were: strong influence of different solvents and poor solubility of complexes in the majority of used solvents. The simplest spectra of free ligands were obtained in d-methanol, but the solubility of L-2 complexes in pure d-methanol was insufficient and finally a mixture of d-methanol and d-chloroform was used.
In the spectrum of free L-1 five broad signals at 2.5–3.6 ppm and four well defined signals at 6.7–7.3 ppm were observed (Fig. 6a). Addition of zinc acetate caused gradual increase of complex signals and diminution of host signals (Fig. 6b). Moreover, in the solution which contains L-1 and zinc acetate in molar ratio 1:2 only complex signals can be recorded (Fig. 6c). However, it was difficult to judge which of nitrogen atom was engaged in complex formation. The observed changes of spectra in the aliphatic region could suggest that at least two of nitrogen atoms were involved in complex formation.

1H NMR spectra of ligand L-1 recorded in d-methanol: (a) free ligand L-1; (b) equilibrium between free ligand L-1 and complexes; (c) complex of L-1 with zinc acetate
The spectra of free ligand L-2 and its complexes with zinc acetate were recorded in the mixture of d-methanol and d-chloroform (7:1.5 v/v) (Fig. 7).

1H NMR spectra of free ligand L-2 and its complex with zinc acetate recorded in the mixture of d-methanol and d-chloroform
Firstly, salt addition caused broadening of all peaks. Formation of the new signals became when 2:1 (L-2:Zn(CH3COO)2) ratio is achieved. Afterwards increase of intensity of complex signals was observed. The most complex spectra were those recorded during complex formation of L-2 with aluminum nitrate. The results are shown in Fig. 8. Gradual salt addition caused significant changes especially in aliphatic region. That might suggest formation of several complexes in solution.

1H NMR spectra of free ligand L-2 and its complex with aluminum nitrate recorded in the mixture of d-methanol and d-chloroform
Fluorescent responses of L-1 and L-2 to various metal ions in acetonitrile solution (excitation at 310 nm for L-1 and 288 nm for L-2) were studied. The emission of L-1 was more or less quenched by Ni2+, Co2+, Cu2+ and Cd2+. On the other hand, Zn2+ enhanced the emission and shifted the emission band. Fluorescence titrations of L-1 were performed for selected cations (Fig. 9). In case of titration of L-1 (7.14 × 10−5 M, λexc = 294 nm) with Cd2+, cations gradual decreasing of emission band was observed (Fig. 9a). After addition of 8.4 × 10−9 mol of Cd(ClO4)2 the emission decreased 10 times. Further titration caused only insignificant changes. Similar results were obtained for other cations, but the addition of just 1 × 10−6 mol of Zn2+effected in increasing emission (L-1 6.63 × 10−5 M). After addition of the 2 × 10−6 mol the emission rose 4 times (Fig. 9b).

Emission spectra recorded in acetonitrile solution containing: (a) ligand L-1 (7.14 × 10−5 M) with cadmium perchlorate (0–3.84 × 10−4 M), (b) ligand L-1 (6.63 × 10−5 M) with zinc perchlorate (0–4.76 × 10−4 M)
Compound L-2 showed different properties. Among studied cations Cd2+ and Zn2+ increased the emission, while Co2+, Ni2+ and Cu2+ completely quenched the emission. Fluorescence titrations of L-2 with Cd2+ and Zn2+ are shown in Fig. 10. In case of titration with Cd2+, the gradual increase of the emission band was observed. After addition of 5 × 10−7 mol of Cd(ClO4)2 the emission increased 2.5 times. In the case of Zn2+ the characteristic band shift and slow increase of emission was observed up to the addition of a 3 × 10−7 mol of Zn(ClO4)2. Increased Zn2+ additions caused significant changes and greatly enhanced the emission (6 times).

Emission spectra recorded in acetonitrile solution containing ligand L-2 (2.52 × 10−5 M, λexc = 283 nm) with (a) cadmium perchlorate (0–1.39 × 10−4 M), (b) zinc perchlorate (0–1.42 × 10−4 M)
Furthermore L-2 showed significant emission enhanced by the presence of Al3+. The major emission enhancement was observed after the addition of 1 equivalent of Al3+ (Fig. 11). Plot of emission versus Al3+: L-2 ratio showed an insignificant increase of the emission band up to molar ratio 1:1 (Al+3: L-2), then a great enhancement in the emission at λ = 420 nm took place. Complexation with Al+3 ions increased tenfold the emission intensity at 420 nm (Fig. 11b).

Emission spectra recorded in methanol solution containing (a) ligand L-2 (2.57 × 10−5 M) with aluminium perchlorate (0–1.86 × 10−4 M), (b) Dependence of fluorescence intensity at 420 nm for ligand L-2 with aluminium perchlorate
Conclusions
The alternative, very selective reaction of monoacylation of unprotected cyclen has been described. The possibility to use the same synthetic pathway for diamide preparation was mentioned. UV–vis and pH titration allowed us to determinate pKa values for both compounds. The obtained results from two different methods were comparable.
L-1 selectively binds zinc cations. The introduction of the second amide group to cyclen considerably changes complexing ability of L-2. Compound L-2 forms complexes with Zn2+, Cd2+, and Al3+ cations. Newly designed amide cyclen derivatives with one or two fluorescing salicylic acid moieties provide novel properties to salicylic fluorescence by itself and by metal complexes. The addition of Ni2+, Co2+, and Cu2+ perchlorates caused quenching of the emission bands of presented ligands, but in contrast to L-1, compound L-2 forms fluorescent complexes not only upon addition of Zn2+, but also Cd2+ and Al3+. The presence of free hydroxyl group strongly influences fluorescence and complexing ability of molecules.
References
Kruppa, M.G., König, B.: Synthesis of chiral amino acids with metal ion chelating side chains from l-serine using Negishi cross-coupling reaction. Tetrahedron 62, 1360–1364 (2006)
Robinson, T.S., Wyness, O., Lincoln, S.F., Taylor, M.R., Tiekink, E.R.T., Wainwright, K.P.: A structural study of the synergic envelopment of acetonitrile by a Cd(II) activated molecular receptor formed from cyclen with appended 2-hydroxy-3-phenylpropyl moieties. Inorg. Chim. Acta 359, 1413–1420 (2006); Gunnlaugsson, T., Davies, R.J.H., Nieuwenhuyzen, M., O’Brien J.E., Stevenson, C.S., Mulready, S.: Synthesis, structural and biological evaluation of GlyAla based lanthanide macrocyclic conjugates as supramolecular ribonuclease mimics. Polyhedron 22, 711–724 (2003)
Hwang, S., Cha, W., Meyerhoff, M.E.: Polymethacrylates with a covalently linked CuII-cyclen complex for the in situ generation of nitric oxide from nitrosothiols in blood. Angew. Chem. Int. Ed. 45, 2745–2748 (2006); Kim, D.H., Han, M.S.: Molecular probe for selective detection of thiols in water of neutral pH. Bioorg. Med. Chem. Lett. 13, 2543–2546 (2003)
Felix, V., Santos, T.M., Madureira, J., Mirante, F., Quintal, S., Goodfellow, B.J., Santana-Marques, M.G., Pedrosa de Jesus J., Drew, M.G.B., Calhorda, M.: Structural characterisation, DFT studies of [Cr(cyclam)(O-dmso)Cl]2+: a new precursor complex towards potential DNA intercalators. J. Inorg. Chim. Acta 356, 335–342 (2003)
Bender, J.A., Meanwell, N.A., Wang, T.: The mono-functionalization of symmetrical polyamines. Tetrahedron 58, 3111–3128 (2002); Li, C., Wong, W.T.: A convenient method for the preparation of mono N-alkylated cyclams and cyclens in high yields. Tetrahedron Lett. 43, 3217–3220 (2002)
Fricke, T., Hamann, J., Bahadir, M., König, B.: Investigation of carrier-mediated anion co-transport through organic membranes by use of competitive transport experiments. Anal. Bioanal. Chem. 374, 148–154 (2002); Skwierawska, A.: Esters of p-Nitrophenol In synthesis of 1-N-Monoamide and 1,7-Bis-N,N′-Amide derivatives of 1,4,7,10-tetraazacyclododecane. Pol. J. Chem. 80, 1353–1361 (2006)
Patinec, V., Yaouanc, J.J., Clement, J.C., Handel, H., des Abbayes, H.: Mono N-Alkylation and N-acylation of cyclen and Cyclam via their metaltricarbonyl complexes (M = Cr, Mo). Tetrahedron Lett. 36, 79–82 (1995); Li, Z., Undheim, K.: Selective mono- and 1,4-Di-N-Alkylations of 1,4,7,10-tetraazacyclododecane. Acta Chem. Scand. 52, 1247–1253 (1998)
Reany, O., Gunnlaugsson, T., Parker, D.: A model system using modulation of lanthanide luminescence to signal Zn2+ in competitive aqueous media. J. Chem. Soc., Perkin Trans. 2, 1819–1831 (2000); Gunnlaugsson, T.: A novel Eu(III)-based luminescent chemosensor: determining pH in a highly acidic environment. Tetrahedron Lett. 42, 8901–8905 (2001)
Subat, M., König, B.: N-Arylation of 1,4,7,10-tetraazacyclododecanes. Synthesis 12, 1818–1825 (2001)
Ritter, S.C., Eiblmaier, M., Michlova, V., König, B.: Chiral NADH model systems functionalized with Zn(II)-cyclen as flavin binding site. Tetrahedron 61, 5241–5251 (2005)
Li, C., Wong, W.T.: A selective one-step synthesis of tris N-alkylated cyclens Tetrahedron 60, 5595–5601 (2004)
Lajunen, L.H.J., Portanova, R., Piispanen, R.J., Tolazz, M.: Critical evaluation of stability constants for alpha-hydroxycarboxylic acid complexes with protons and metal ions and the accompanying enthalpy changes Part I: Aromatic ortho-hydroxycarboxylic acids. Pure Appl. Chem. 69(2), 329–382 (1997); Surányi, T.M., Djurendic, E.A., Vastag, G.G.: Spectrophotometric study of the formation of Iron(III) complexes with some salicylic acid derivatives. Collec. Czech. Chem. Commun. 60(3), 464–472 (1995)
Skwierawska, A., Paluszkiewicz, E., Przyborowska, M., Ossowski, T.: The synthesis of 1,4,7,10-tetraazacyclododecane with Acetyl. J. Inc. Phenom. Macrocyc. Chem. 49, 21–26 (2004)
Skwierawska, A., Paluszkiewicz, E.: High yield synthesis and preliminary spectroscopic study of mono-N-alkylated cyclen derivatives of salicylic acid. J. Inc. Phenom. Macrocyc. Chem. 56, 323–330 (2006)
Kostrowicki, J., Liwo, A.: Determination of equilibrium parameters by minimization of an extended sum of squares. Talanta 37, 645–650 (1990)
Cevasco, G., Gunanti, G., Hopkins, A.R., Thea, S., Williams, A.: A novel dissociative mechanism in acyl group transfer from aryl 4-hydroxybenzoates in aqueous solvents. J. Org. Chem. 50, 479–484 (1985)
Oxford Diffraction CrysAlis CCD and CrysAlis RED. Version 1.171, Oxford Diffraction Ltd, Abingdon, Oxfordshire, England (2005)
Sheldrick, G.M. SHELXS97 and SHELXL97, University of Göttingen, Germany (1997)
Farrugia, L.J.: ORTEP-3 for Windows—a version of ORTEP-III with a Graphical User Interface (GUI) J. Appl. Cryst. 30, 565 (1997)
Farrugia, L.J.: J. WinGX suite for small-molecule single-crystal crystallography. Appl. Cryst. 32, 837–838 (1999)
Hahn, T. (ed.): International tables for crystallography, vol. A, Kluwer Academic Publishers, Dordrecht (2002)
Acknowledgements
Financial support from Gdańsk University of Technology (DS 014668/003 and BW 014694/054) is gratefully acknowledged. We thank Dr. J. Chojnacki from Gdańsk University of Technology for X-ray crystal structure determination of L-2.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Skwierawska, A., Paluszkiewicz, E., Przyborowska, M. et al. 1,4,7,10-Tetraazacyclododecane incorporating salicylic acid moieties synthesis and properties. J Incl Phenom Macrocycl Chem 61, 305–312 (2008). https://doi.org/10.1007/s10847-008-9423-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10847-008-9423-7