
Abstract
Maintaining pure cultures using preservation methods is of high importance for biotechnological purposes. However, preservation does not necessarily guarantee the genetic stability of these cultures. Therefore, preservation methods are currently needed to assure viability as well as genetic, physiological, and morphological integrity across storage periods. In this study, preservation of five isolates from the microalgae and cyanobacteria collection of the Plant Biology Department, Federal University of Viçosa, Minas Gerais, Brazil was investigated via monthly analyses of cell viability, biomass recovery, and contaminant concentrations over a period of 120 days. Lyophilization was adequate for both heterocystous cyanobacteria and other strains that were able to differentiate hormogones or to synthesize thick layers of exopolysaccharides. Lyophilization was also able to maintain cultures with low levels of contaminants. Dimethyl sulfoxide was relatively efficient, though some of the strains were susceptible to its cytotoxic effects. Our results demonstrated that cryopreservation with glycerol was the most efficient method. The ability to routinely preserve cyanobacterial strains reduces costs associated with maintaining large culture collections and reduces the risks of losing particular strains or species through contamination and genetic drift. The results obtained in this study are therefore discussed in the context of the efficiency of the methods and the current need to develop suitable methods for maintenance of cyanobacterial collections.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cyanobacteria are among the oldest organisms on earth and have played a crucial role in the evolution of life by generating both an oxidizing atmosphere via the production of oxygen as a photosynthetic byproduct and organic carbon, which is the major ecological energy commodity. These organisms constitute a unique phylogenetic group of bacteria that evolved approximately three billion years ago and were the first organisms to perform oxygenic photosynthesis (Hackenberg et al. 2011). In addition, cyanobacteria occupy diverse ecological niches and exhibit enormous diversity in terms of their habitats, physiology, morphology, and metabolic capabilities (Beck et al. 2012). In fact, cyanobacteria appear to be able to establish competitive growth in almost any environment where there is, at least temporarily, liquid water and sunlight (Badger et al. 2006).
Cyanobacteria can be developed as a highly productive microbial cell factory that can harvest solar energy and convert atmospheric CO2 to useful products (Parmar et al. 2011). Accordingly, cyanobacteria are advantageous organisms for use in industrial applications, as they exhibit rapid cell growth, have simple nutrient requirements (mainly water, sunlight, and CO2) (Ruffing 2011), and are naturally transformable, thus presenting the potential to be genetically engineered (Golden et al. 1987; Koksharova and Wolk 2002; Huang et al. 2010; Heidorn et al. 2011; Ruffing 2011; Wilde and Dienst 2011).
Unlike most other groups of microorganisms, both eukaryotic microalgae and prokaryotic cyanobacteria (blue-green algae) have traditionally been maintained via serial subculturing, with the frequency of transfer being largely determined by the growth characteristics of the strain (Day and DeVille 1995; Day 2007). However, continuous maintenance of large culture collections and actively growing cyanobacterial strains over long periods of time is relatively complex, time consuming, and when it involves large numbers of cultures, expensive. This technique has the disadvantages of being labor intensive and being characterized by a constant risk of strains or species becoming contaminated and of genetic drift occurring under serial subculturing (Lorenz et al. 2005; Rhodes et al. 2006). The ability to routinely preserve cyanobacteria would circumvent some of these issues and enable a larger number of strains to be maintained.
Cyanobacteria preservation methods can at least theoretically be divided in two groups according to maintaining strains in either continued or suspended metabolism. The methods that allow continued metabolism include storage in sterile water, subculturing in agar, cool storage at temperatures ranging from 5 to 8 °C and deep freezing at −20 °C. Preservation via suspended metabolism is achieved by drying cultures using silica gel or lyophilization, or through rapid snap freezing with liquid nitrogen (Borman et al. 2006).
Lyophilization preservation has been successfully applied for preserving yeast and sporulating fungi as well as bacteria (Berny and Hennebert 1991). The advantages of this methodology include protection from contamination or infestation during storage, long-term viability, and the facility of strain distribution (Smith and Onion 1983). However, not all strains survive the process and, among those surviving, very low quantitative viability levels have been extensively reported (Atkin et al. 1949; Kirsop 1955; Smith and Onion 1983; Berny and Hennebert 1991; Bunse and Steigleder 1991).
The application of cryopreservation has stimulated several studies addressing the effects of low temperatures and freezing processes on cells and tissues in order to develop efficient protocols for genotype preservation (Day and DeVille 1995; Mycock et al. 1995; Newton et al. 1998; Day 2007). Accordingly, it has been widely assumed that for cryopreservation methods to be successful, the addition of an intracellular cryoprotective agent in the freezing solution is indispensable (Yu and Quinn 1994; Smith et al. 2001; Castro et al. 2011). However, issues concerning the intracellular cryoprotective agents used (e.g., their metabolism and potential toxicity) must be examined carefully in order to select the most suitable cryoprotectant for a specific structure. Although cryopreservation has been thought of as the most suitable method for achieving long-term genetic stability (Grout 1995), this technique has not been adopted by a large number of researchers (Day et al. 2000; Day 2007). Thus, although few cyanobacterial cryopreservation studies have been previously carried out (Watanabe 1959; Holm-Hansen 1973; Box 1988), a cryopreservation protocol appropriate for a wide range of cyanobacterial strains has not yet been established. The aim of this research initiative was to determine the optimal system for the cryopreservation of morphologically distinct cyanobacterial strains using different cryoprotective agents. The effects of the different methods applied were monitored, and the combined data obtained indicate an alternative preservation method for cyanobacterial collections.
Materials and methods
The five cyanobacterial strains used in this study were selected from the microalgae and cyanobacteria collection of the Plant Biology Department, Federal University of Viçosa, Minas Gerais, Brazil by choosing morphological representatives of four orders within the phylum Cyanobacteria: Chroococcales (Synechocystis sp.), Oscillatoriales (Phormidium sp.), Nostocales (Brasilonema octagenarum and Nostoc sp.), and Stigonematales (Stigonema sp.) (Anagnostidis and Komárek 1985). These five strains represent a variety of different cell morphologies, such as, unicellular forms, motile and nonmotile cells, filamentous forms with different types of cells (vegetative cells, heterocysts, and akinetes), and non-heterocystous forms (Table 1; Fig. 1).

Morphological characteristics of the selected strains under control conditions and after preservation. The tested strains were Synechocystis sp. (a–c), Phormidium sp. (d–f), B. octagenarum (g–i), Nostoc sp. (j–l), and Stigonema sp. (m–o). Vegetative cells and different cellular structures were observed after 12 h of incubation in Ringer’s solution containing 0.2 % TTC; the presence of purple formazan crystals indicates a positive reaction. Control cultures are shown in the first column, lyophilized cultures in the second column, and cultures cryopreserved with glycerol in the third. Abbreviations: colony (co); vegetative cells (vc); vegetative cells in the differentiation stage (vc 2 ); filament (f); hormocysts (hc); hormogones (hm); heterocysts (ht); false branching (fb); true branching (tb)
For each strain, aliquots of 0.5 mL were taken from cultures and inoculated into 125-mL Erlenmeyer flasks containing 50 mL of BG 11 culture medium (Rippka et al. 1979), with 0.15 % (w/v) NaNO3 being added only to the cultures of non-heterocystous cyanobacteria. The Erlenmeyer flasks were maintained under an 18/6-h day/night light cycle with a 50-μmol photons m−2 s−1 light intensity, a temperature of 24 ± 2 °C, and constant shaking (90 rpm) for 60 days to allow differentiation of structures such as akinetes, hormogones and hormocysts, given that these structures maximize the tolerance of the strains to cryopreservation conditions and lyophilization.
Sample preparation
The cultures were subjected to homogenization and centrifuged at 15,000×g for 15 min. The supernatant was discarded, and the pellet was resuspended in 5 mL of saline solution (NaCl 0.85 % w/v). This procedure was repeated twice to remove biomass residues. The final pellet was resuspended in BG 11 medium (with or without NO3 −).
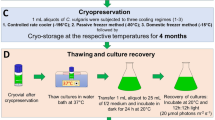
Maintenance by cryopreservation
For the experiments involving cryopreservation, dimethyl sulfoxide (DMSO) and glycerol were used as cryoprotective agents at concentrations of 5 % and 10 % v/v, respectively (Romo and Becares 1992; Simione 1998; Smith et al. 2001). The cryoprotectants were diluted in culture medium to ensure more uniform exposure when added to the cell suspension.
Two aliquots of the same volume were removed from homogenized cultures and centrifuged, and the supernatant was discarded. A solution of fresh culture medium containing one of the cryoprotective agents (DMSO or glycerol) was added to each pellet. The equilibration period for the cultures was equal to 15 min (Simione 1998; Day 2007). After 10 min of exposure, aliquots of 1 mL were transferred to labeled 1.5 mL tubes (strain/method), and when the equilibration period was complete, the 1.5-mL tubes were transferred to an ultrafreezer (−80 °C) and stored in a vertical orientation.
Maintenance by lyophilization
Aliquots of 1.0 mL were transferred to labeled 1.5-mL tubes (strain/method), which were vertically oriented prior to being subjected to flash freezing using liquid nitrogen (Smith et al. 2001; Day 2007). After freezing, the 1.5-mL tubes were placed in a model L101 lyophilizer (LIOTOP, Brazil), and the samples were dried completely. After this process was complete, all materials were kept in a desiccator containing blue silica gel.
Viability analyses and recovery time for biomass
The viability level and capacity to produce biomass were monitored monthly over a period of 120 days in the five strains subjected to cryopreservation and lyophilization. Frozen samples were removed each month from the stored samples and thawed in a Becker containing sterile saline solution at approximately 25 °C. The content of each 1.5-mL tube was transferred to centrifuge tubes containing 9 mL of saline solution to dilute the added cryoprotectant. The cultures were centrifuged, and the supernatant was discarded. This procedure was repeated twice to completely remove the cryoprotectant (Simione 1998; Day 2007). The lyophilized material was then removed from the desiccator, rehydrated in 5 mL of saline solution, and homogenized in a shaker tube. The cell suspension was centrifuged, and the pellet was resuspended in 1 mL of saline solution.
To evaluate the biomass production capacity, 200 μL of a thawed or rehydrated cell suspension was inoculated into Erlenmeyer flasks containing 20 mL of BG 11 culture medium and maintained at room temperature under the photoautotrophic conditions described previously. The evaluation of growth was performed by daily observation to visualize the formation of colonies inside the Erlenmeyer flasks (Taylor and Fletcher 1998; Day 2007).
The identification of viable cells was performed by adding 0.7 mL of Ringer’s solution containing 0.2 % TTC to 1 mL of a cell suspension. The reaction was maintained in the dark for 12 h (Markelova et al. 2000; Marin et al. 2006), and viable cells were determined based on the occurrence of purple coloration (Markelova et al. 2000). Viability levels were determined via cell counting performed in Neubauer chamber (Optik Labor, Germany) and were calculated as follows:
Quantification of contaminants
The presence of contaminants (heterotrophic bacteria adhered to exopolysaccharides) in both control cultures and preserved samples was quantified monthly over a 120-day period to assess whether significant changes in the number of the contaminant bacteria occurred due to the different methodologies used.
To obtain suspensions of bacterial cells, cultures of each strain were subjected to exopolysaccharide extraction according to Fiore and colleagues (2000). After this procedure, serial dilutions were performed, and a 25-μL aliquot was spread on nutrient agar plates. The plates were maintained at 30 °C for 12–24 h, or until colonies appeared to quantify the number of colony-forming units (CFU) (Gerhardt and Drew 1994).
Statistical analyses
The viability and contaminant analyses were performed using a completely randomized design with three replicates under a 3 × 4 factorial design based on the three methods of preservation (cryopreservation using DMSO or glycerol and lyophilization) and four time intervals (30, 60, 90, and 120 days). The obtained data were analyzed with the SAEG software, version 9.1 (SAEG 2007) via analysis of variance and subsequent comparison of means using the Dunnett test at the 5 % significance level. Additionally, evaluation of the effect of time on each method was performed by linear regression.
Results
Viability analyses
Prior to cryopreservation, all five cyanobacterial strains were analyzed using light microscopy to confirm the presence of morphological characteristics intrinsic to the selected strains (Table 1; Fig. 1). The strains used in the present study for evaluation of the preservation methods showed high viability levels (from 78 % to 98 %) as well as differentiated structures after 60 days of cultivation. In general, the highest viability (86.6 %) was observed when using glycerol as a cryoprotective agent. When DMSO was used as a cryoprotective agent, a cell viability of 64 % was maintained after 120 day of cultivation. Lyophilization preservation resulted in a very low percentage number of viable cells (14 %).
Cryopreservation in the presence of glycerol
Cryopreservation of Synechocystis sp. in the presence of glycerol led to a viability level reduction of nearly 10 % after 120 days of cultivation (Fig. 2a), with a significant interaction detected between the investigated variables of time and method. Utilization of glycerol as a cryoprotectant was proven to be more efficient than the other methods for the cryopreservation of Phormidium sp. (Fig. 2b), Nostoc sp. (Fig. 2d), and Stigonema sp. (Fig. 2e), with a constant high viability of cells observed throughout the incubation period (120 days). Brasilonema octagenarum cells showed the highest viability level at the beginning of the treatment; however, during the preservation period, a significant reduction of the viability level was detected (Fig. 2c).

Viability levels of the cyanobacterial strains. Synechocystis sp. (a), Phormidium sp. (b), B. octagenarum (c), Nostoc sp. (d), and Stigonema sp. (e) subjected to lyophilization and cryopreservation with glycerol and DMSO. Linear regression analyses was realized to assess the possible interaction effect between the variables time (days) and method. The equation is shown in each panel

Presence of contaminants in each strain is affected by the applied preservation methods. The colony-forming unit (CFU mL−1) values were transformed to logarithmic values. The tested strains used were Synechocystis sp. (a), Phormidium sp. (b), B. octagenarum (c), Nostoc sp. (d), and Stigonema sp. (e), which were subjected to lyophilization and cryopreservation with glycerol and DMSO. An asterisk indicates values that were judged to be significantly different from the control (P < 0.05) using the Dunnett test
Cryopreservation in the presence of DMSO
When the cyanobacteria were cryopreserved in the presence of DMSO, Synechocystis sp. showed the highest viability level. A significant interaction between the time of cryopreservation and the method used was also observed (Fig. 2a). Although a similar pattern was observed for Phormidium sp., B. octagenarum and Stigonema sp., this method did not provide the highest viability levels for these strains (Fig. 2b, c, e). Less than 6 % of cells were observed to be viable throughout the preservation period for Nostoc sp. (Fig. 2d).
Lyophilization
No positive reaction with TTC was observed in any of the lyophilized cells of Synechocystis sp., Phormidium sp., and B. octagenarum (Fig. 2a, b, c), whereas approximately 4.0 % of the Nostoc sp. cells were able to reduce TTC, resulting in purple crystal formation, and this percentage was maintained throughout the evaluated preservation period (Fig. 2d). Lyophilized cells of Stigonema sp. showed a positive reaction level of greater than 70 % at 30 days of cultivation, though this value was significantly reduced during the preservation period (Fig. 2e).
Stained cell types
Given that some non-heterocystous cyanobacteria, such as Synechocystis sp., do not exhibit differentiated cell structures, and vegetative cells are more susceptible to lyophilization, a positive reaction was only observed in vegetative cells in both control and cryopreserved cultures (Fig. 1a, b, c). Although both vegetative cells and hormogones of Phormidium sp. and B. octagenarum reacted with TTC in normal and cryopreserved cultures (Fig. 1f, i), rehydration was not sufficient to rescue the metabolic activity of these strains (Fig. 1e, h).
Few hormogones of cryopreserved cultures of Nostoc sp. showed a positive reaction, which was mainly observed in cells maintained with glycerol but also with DMSO (Fig. 1l). It is important to note that vegetative cells with positive reaction were mainly located in the interior of the colony, particularly in colonies preserved by lyophilization or frozen with DMSO (Fig. 1k).
Interestingly, both vegetative cells and hormocysts of Stigonema sp. exhibited a positive reaction with TTC under all of the tested methods. However, in lyophilized samples of young vegetative cells, there was no positive reaction detected. It was also possible to observe that the distinct cell types presented different sizes of the crystals (Fig. 1n, o).
Recovery time of biomass
To obtain a clear reflection of the tested methods, biomass recovery was performed after cryopreservation and lyophilization. Biomass production was observed after 2 days in Synechocystis sp. cryopreserved in the presence of DMSO but not until after 5 days for the cells preserved with glycerol (Table 2). In sharp contrast, no growth was observed for lyophilized cells of Synechocystis sp. (Table 2).
The cells of Phormidium sp. maintained under cryopreservation produced biomass after 2 days of cultivation using either glycerol or DMSO, whereas lyophilized cells were only able to form new colonies at the first and third sampling after 7 and 12 days of evaluation, respectively (Table 2).
For B. octagenarum, 10 and 22 days were necessary for biomass production to occur in cryopreserved and lyophilized cells, respectively. Nostoc sp. cells were able to form colonies after 4, 7, and 10 days when cryopreservation was performed with glycerol or DMSO or when lyophilization was used, respectively. Additionally, in Stigonema sp. cells cryopreserved with glycerol or DMSO, biomass growth was evident after 5 and 7 days of cultivation, respectively, whereas lyophilized cells were only able to generate colonies after 13 days during the first 2 months and after 20 days in the subsequent evaluations (Table 2).
Quantification of contaminants
To obtain corroborative evidence for the results highlighted above, we assessed the possibility of contamination under each of the evaluated preservation methods based on determination of the presence of undesired organisms by quantifying the CFU number (Fig. 3). When the contamination present in control cells was compared with that in lyophilized cells, we observed a reduction of contaminants by more than 99 % after preservation. In addition, no difference was observed when comparing cryopreservation in the presence of DMSO or glycerol in any of the strains analyzed in this study (Table 3).
Discussion
The superior photosynthesis capabilities of cyanobacteria allow them to convert up to 10 % of the sun’s energy into biomass, compared to the 1 % converted by conventional energy crops such as corn or sugarcane, or the 5 % achieved by algae (Parmar et al. 2011; Jones and Mayfield 2012). Both cyanobacterial and microalgal systems could contribute to sustainable bioenergy production. However, different biotechnical, environmental, and economic challenges have to be overcome before energy products from these systems can enter the market (Parmar et al. 2011). One of these challenges is clearly the development of a suitable method for the preservation of cyanobacterial strains to prevent the accumulation of mutations resulting not only in morphological but also in undesired biochemical alterations (Borman et al. 2006). Here, we have developed a new system allowing rapid quantification of cell viability after preservation. The parameter used for this purpose in the present study provides useful information about the metabolic state of single cells after freezing and rehydration. Thus, even in the absence of a positive reaction with TTC, the cells were able to reactivate their metabolism and produce biomass.
Utilization of glycerol as a cryoprotective agent was efficient, as high viability levels were maintained (Phormidium sp., Nostoc sp., and Stigonema sp.), or the viability was only slightly decreased (B. octagenarum) during the storage period. Although this cryoprotectant was not as efficient as DMSO for the preservation of Synechocystis sp., the viability levels observed in the presence of glycerol were significantly higher. While the mechanism underlying this response is currently unclear, it is tempting to speculate that because glycerol is a constituent of cellular membranes, it can be accumulated and metabolized and does therefore not induce cytotoxic effects, as observed previously (Mackay et al. 1984; Hagemann et al. 1997; Marin et al. 2006). In a similar vein, it might act to protect cells during storage under low temperatures, avoiding intracellular ice formation as well as possible damage due to dehydration (Han et al. 2009).
The observation that cryopreservation in the presence of DMSO was very efficient for Phormidium sp., B. octagenarum, Stigonema sp. and, particularly, Synechocystis sp. is highly intriguing, especially in light of the fact that we were able to demonstrate rapid biomass recovery in these strains. Several reports have suggested that characteristics of DMSO such as its easy diffusion through cellular membranes, coupled with its ability to efficiently interact with phospholipids, resulting in the maintenance of membrane permeability during freezing (Sojka et al. 1990; Yu and Quinn 1994; Aye et al. 2010; Castro et al. 2011), would at least in part explain such results. However, it is important to mention that although for both Phormidium sp. and B. octagenarum, the use of DMSO promoted relatively higher viability levels, these levels were slightly lower than those measured in the presence of glycerol. Additionally, the low cell viability observed for Nostoc cf. carneum is highly suggestive of the occurrence of cytotoxic effects of this cryoprotective agent (Yu and Quinn 1994; Aye et al. 2010) under the concentration used (5 %), which might have been excessive for this particular strain. Intriguingly, even though a very low percentage of positive reactions were observed, biomass production was maximal, most likely due to the presence of hormogones and akinetes.
The total absence of a positive reaction with TTC in lyophilized cells of B. octagenarum, Phormidium sp., and Synechocystis sp. is highly consistent with the reduced activity of important metabolic process such as photosynthesis, respiration, and nitrogen assimilation under these conditions, limiting the availability and activity of NADH and NADPH dehydrogenases that are able to reduce TTC to formazan (Beloti et al. 1999; Mohammadzadeh et al. 2006). It is, however, surprising that even in the absence of the formation of purple crystals, B. octagenarum and Phormidium sp. were able to somewhat reestablish their metabolism, as observed by the biomass production (Table 2), which is an indicator parameter that has been used previously (Bezerra et al. 2006; Silva et al. 2007). Although the survival of these strains can be mainly associated with the presence of hormogones (Fig. 1d, h), to elucidate the mechanism(s) by which this survival is achieved, further experiments will clearly be required.
The intriguing lack of biomass production in lyophilized cells of Synechocystis sp. could be related to the higher sensitivity of vegetative cells to vacuum dehydration and is in keeping with the hypothesis that this process leads to alterations of membrane permeability and the loss of cell metabolites (Cordero and Voltolina 1997; Tan 1997). However, it could also be associated with the absence of differentiation of hormogones or other cell structures during the life cycle of this strain (Nakao et al. 2010; Komárek and Hauer 2011). In good agreement with this hypothesis of a high sensitivity of vegetative cells to lyophilization, we also found that only Phormidium sp. exhibited an increase in biomass in two out of four evaluations. Notwithstanding the thick layers of exopolysaccharides observed in this strain coupled with the differentiated hormogones might play an important role in the survival of a few cells after lyophilization (Hill et al. 1994; Nicolaus et al. 1999). Accordingly, it has previously been demonstrated that non-heterocystous organisms are more sensitive to lyophilization (Corbett and Parker 1976).
One conspicuous feature of the results presented here is the complete recuperation of lyophilized cells of B. octagenarum, which is suggestive of inherent characteristics of this genus. All three strains of this genus are found in subaerophytic habitats, and they are able to colonize eucalyptus leaves, bromeliads, bark trees and stone surfaces, among other substrates (Fiore et al. 2007; Aguiar et al. 2008; Sant'Anna et al. 2011). Thus, it is highly possible that they are more often subjected to variations in relative humidity or limited water availability, suggesting that the filaments and hormogones of B. octagenarum are tolerant to desiccation. While the precise nature of these conditions could not be fully resolved in this study, it remains an exciting topic for future research.
Only in Stigonema sp. positive reactions of differentiated structures with TTC were observed after the rehydration of the samples. The smaller crystal size observed in the interior of hormocysts indicated that these structures have a relatively low metabolism (Fig. 1n). When taken together, these data suggest that, by contrast to the situation observed for hormogones and akinetes, hormocysts are able to retain an active metabolism, even under conditions of extreme desiccation. Additionally, the absence of a positive reaction in younger vegetative cells indicates a stronger impact of the dehydration process on this cell type (Cordero and Voltolina 1997; Tan 1997). The longer time interval necessary for colony formation in these cultures (13 to 20 days; Table 2) can therefore be explained by the reduced metabolic activity of hormocysts as well as the low number of viable vegetative cells.
Not surprisingly, the applied cryopreservation methods did not alter the initial number of contaminants throughout the storage period. This shows that these methods are most likely not only efficient for cyanobacteria preservation but also for preserving some of the heterotrophic bacteria adhered to the exopolysaccharides. Although after lyophilization, the contamination observed represented less than 1 % of that observed in both control and cryopreserved samples, it is assumed that most of the surviving bacteria may exhibit poor viability due to factors associated either with inadequate drying (Smith et al. 2001) or, most likely, differences in the cell wall composition. Taken together, these results suggest that the contaminants present in these cultures are composed primarily of Gram-negative bacteria, as it has been demonstrated (Miyamoto-Shinohara et al. 2000; Nocker et al. 2012) that these strains are more susceptible to drying than the Gram-positive strains.
In summary, we have presented compelling evidence that cryopreservation is a valuable tool for cyanobacterial preservation, providing support for future biotechnology research as well as allowing safeguarding of genetic material and facilitating its propagation. However, the success of this procedure depends on the proper use of a cryoprotective agent which although essential, can cause irreversible cell damage that can result in cellular death. Our data indicated that cryopreservation in the presence of glycerol (10 %) was generally the method with the highest efficiency for the tested strains. The results obtained following cryopreservation in the presence of DMSO are not only confirmatory regarding its described efficiency as a cryoprotectant but also demonstrated that, even at low concentrations, this agent can have cytotoxic effects. The lyophilization method has been proven to be suitable for the preservation of heterocystous cyanobacteria (order Nostocales) as well as for preventing contamination, although due to the lower survival of strains that are unable to differentiate hormogones and/or to synthesize thick layers of exopolysaccharides, improvement of this method is clearly required. In conclusion, although there is no ideal cryoprotective agent that is both able to completely protect cells at low temperatures and is free of toxicity, the experiments conducted here allowed us to demonstrate that the currently employed cryoprotectants enable the preservation of cyanobacterial strains and that the selection of the cryoprotectant to be used is highly dependent on the strain. It will therefore be important to expand this approach to a greater number of the strains maintained in cyanobacterial collections.
References
Aguiar R, Fiore MF, Franco MW, Ventrella MC, Lorenzi AS, Vanetti CA, Alfenas AC (2008) A novel epiphytic cyanobacterial species from the genus Brasilonema causing damage to Eucalyptus leaves. J Phycol 44:1322–1334
Anagnostidis K, Komárek Jí (1985) Modern approach to the classification system of cyanophytes. 1 - Introduction. Arch Hydrobiol 38–39:291–302
Atkin L, Moses W, Gray PP (1949) The preservation of yeast culture by lyophilization. J Bacteriol 57:575–578
Aye M, Di Giorgio C, De Mo M, Botta A, Perrin J, Courbiere B (2010) Assessment of the genotoxicity of three cryoprotectants used for human oocyte vitrification: dimethyl sulfoxide, ethylene glycol and propylene glycol. Food Chem Toxicol 48:1905–1912
Badger MR, Price GD, Long BM, Woodger FJ (2006) The environmental plasticity and ecological genomics of the cyanobacterial CO2 concentrating mechanism. J Exp Bot 57:249–265
Beck C, Knoop H, Axmann I, Steuer R (2012) The diversity of cyanobacterial metabolism: genome analysis of multiple phototrophic microorganisms. BMC Genomics 13(1):56
Beloti V, Barros MAF, de Freitas JC, Nero LA, de Souza JA, Santana EHW, Franco B (1999) Frequency of 2,3,5-triphenyltetrazolium chloride (TTC) non-reducing bacteria in pasteurized milk. Rev Microbiol 30:137–140
Berny JF, Hennebert GL (1991) Viability and stability of yeast cells and filamentous fungus spores during freeze-drying: effects of protectants and cooling rates. Mycologia 83:805–815
Bezerra CCF, de Lima RF, Lazera MS, Wanke B, Borba CM (2006) Viability and molecular authentication of Coccidioides immitis strains from Culture Collection of the Instituto Oswaldo Cruz, Rio de Janeiro, Brazil. Rev Soc Bras Med Trop 39(3):241–244
Borman AM, Szekely A, Campbell CK, Johnson EM (2006) Evaluation of the viability of pathogenic filamentous fungi after prolonged storage in sterile water and review of recent published studies on storage methods. Mycopathologia 161:361–368
Box JD (1988) Cryopreservation of the blue-green alga Microcystis aeruginosa. Br Phycol J 23:385–386
Bunse T, Steigleder GK (1991) The preservation of fungal cultures by lyophilization. Mycoses 34:173–176
Castro SV, Carvalho AA, Silva CMG, Faustino LR, Figueiredo JR, Rodrigues APR (2011) Intracellular cryoprotectant agents: characteristics and use of ovarian tissue and oocyte cryopreservation. Acta Sci Vet 39:957
Corbett LL, Parker DL (1976) Viability of lyophilized cyanobacteria (blue-green algae). Appl Environ Microbiol 32:777–780
Cordero B, Voltolina D (1997) Viability of mass algal cultures preserved by freezing and freeze-drying. Aquac Eng 16:205–211
Day JG (2007) Cryopreservation of microalgae and cyanobacteria. In: Day JG, Stacey GN (eds) Cryopreservation and freeze-drying protocols vol 368. Methods in molecular biology. Humana Press, New York, pp 141–151
Day JG, DeVille MM (1995) Cryopreservation of algae. In: Day JG, McLellan MR (eds) Methods in molecular biology, vol 38. Methods in molecular biology. Humana Press, New York, pp 81–89
Day JG, Fleck RA, Benson EE (2000) Cryopreservation-recalcitrance in microalgae: novel approaches to identify and avoid cryo-injury. J Appl Phycol 12:369–377
Fiore MF, Moon DH, Tsai SM, Lee H, Trevors JT (2000) Miniprep DNA isolation from unicellular and filamentous cyanobacteria. J Microbiol Meth 39:159–169
Fiore MF, Sant'Anna CL, Azevedo MTP, Komarek J, Kastovsky J, Sulek J, Lorenzi AS (2007) The cyanobacterial genus Brasilonema, gen. nov., a molecular and phenotypic evaluation. J Phycol 43:789–798
Gerhardt P, Drew SW (1994) Liquid culture. In: Gerhardt P, Murray RGE, Wood WA, Kraig NR (eds) Methods for general and molecular bacteriology. American Society for Microbiology, Washington DC, pp 224–247
Golden SS, Brusslan J, Haselkorn R (1987) Genetic engineering of the cyanobacterial chromosome. Methods Enzymol 153:215–231
Grout BWW (1995) Introduction to the in vitro preservation of plant cells, tissues and organs. In: Grout BWW (ed) Genetic preservation of plant cells in vitro. Springer Verlag, Berlin, pp 1–20
Hackenberg C, Kern R, Hüge J, Stal LJ, Tsuji Y, Kopka J, Shiraiwa Y, Bauwe H, Hagemann M (2011) Cyanobacterial lactate oxidases serve as essential partners in N2 fixation and evolved into photorespiratory glycolate oxidases in plants. Plant Cell 23:2978–2990
Hagemann M, Schoor A, Jeanjean R, Zuther E, Joset F (1997) The stpA gene from Synechocystis sp. strain PCC 6803 encodes the glucosylglycerol-phosphate phosphatase involved in cyanobacterial osmotic response to salt shock. J Bacteriol 179:1727–1733
Han X, Ma L, Benson J, Brown A, Critser JK (2009) Measurement of the apparent diffusivity of ethylene glycol in mouse ovaries through rapid MRI and theoretical investigation of cryoprotectant perfusion procedures. Cryobiology 58:298–302
Heidorn T, Camsund D, Huang H-H, Lindberg P, Oliveira P, Stensjo K, Lindblad P (2011) Synthetic biology in cyanobacteria: engineering and analyzing novel functions. Methods Enzymol 497:539–579
Hill DR, Hladun SL, Scherer S, Potts M (1994) Water stress proteins of Nostoc commune (Cyanobacteria) are secreted with UV-A/B-absorbing pigments and associate with 1,4-beta-d-xylanxylanohydrolase activity. J Biol Chem 269:7726–7734
Holm-Hansen O (1973) Preservation by freezing and freeze drying. In: Stein JR (ed) Handbook of phycological methods: culture methods and growth measurements. Cambridge University Press, Cambridge, pp 195–205
Huang H-H, Camsund D, Lindblad P, Heidorn T (2010) Design and characterization of molecular tools for a synthetic biology approach towards developing cyanobacterial biotechnology. Nucleic Acids Res 38:2577–2593
Jones CS, Mayfield SP (2012) Algae biofuels: versatility for the future of bioenergy. Curr Opin Biotechnol 23:346–351
Kirsop B (1955) Maintenance of yeasts by freeze-drying. J Inst Brew 21:466–471
Koksharova OA, Wolk CP (2002) Genetic tools for cyanobacteria. Appl Microbiol Biotechnol 58:123–137
Komárek J, Hauer T (2011) CyanoDB.cz - On-line database of cyanobacterial genera. Word-wide electronic publication, Univ of South Bohemia & Inst of Botany AS CR, http://wwwcyanodbcz
Lorenz M, Friedl T, Day JG (2005) Perpetual maintenance of actively metabolizing microalgal cultures. In: Andersen RA (ed) Algal culturing techniques. Academic Press, New York, pp 145–156
Mackay MA, Norton RS, Borowitzka LJ (1984) Organic osmoregulatory solutes in cyanobacteria. J Gen Microbiol 130:2177–2191
Marin K, Stirnberg M, Eisenhut M, Kraemer R, Hagemann M (2006) Osmotic stress in Synechocystis sp PCC 6803: low tolerance towards nonionic osmotic stress results from lacking activation of glucosylglycerol accumulation. Microbiology 152:2023–2030
Markelova AG, Vladimirova MG, Kuptsova ES (2000) A comparison of cytochemical methods for the rapid evaluation of microalgal viability. Russ J Plant Physiol 47:815–819
Miyamoto-Shinohara Y, Imaizumi T, Sukenobe J, Murakami Y, Kawamura S, Komatsu Y (2000) Survival rate of microbes after freeze-drying and long-term storage. Cryobiology 41:251–255
Mohammadzadeh A, Farnia P, Ghazvini K, Behdani M, Rashed T, Ghanaat J (2006) Rapid and low-cost colorimetric method using 2,3,5-triphenyltetrazolium chloride for detection of multidrug-resistant Mycobacterium tuberculosis. J Med Microbiol 55:1657–1659
Mycock DJ, Wesley-Smith J, Berjak P (1995) Cryopreservation of somatic embryos of four species with and without cryoprotectant pre-treatment. Ann Bot 75:331–336
Nakao M, Okamoto S, Kohara M, Fujishiro T, Fujisawa T, Sato S, Tabata S, Kaneko T, Nakamura Y (2010) CyanoBase: the cyanobacteria genome database update 2010. Nucleic Acids Res 38:D379–D381
Newton H, Fisher J, Arnold JRP, Pegg DE, Faddy MJ, Gosden RG (1998) Permeation of human ovarian tissue with cryoprotective agents in preparation for cryopreservation. Hum Reprod 13:376–380
Nicolaus B, Panico A, Lama L, Romano I, Manca MC, De Giulio A, Gambacorta A (1999) Chemical composition and production of exopolysaccharides from representative members of heterocystous and non-heterocystous cyanobacteria. Phytochemistry 52:639–647
Nocker A, Fernández PS, Montijn R, Schuren F (2012) Effect of air drying on bacterial viability: a multiparameter viability assessment. J Microbiol Meth 90:86–95
Parmar A, Singh NK, Pandey A, Gnansounou E, Madamwar D (2011) Cyanobacteria and microalgae: a positive prospect for biofuels. Bioresour Technol 102:10163–10172
Rhodes L, Smith J, Tervit R, Roberts R, Adamson J, Adams S, Decker M (2006) Cryopreservation of economically valuable marine micro-algae in the classes Bacillariophyceae, Chlorophyceae, Cyanophyceae, Dinophyceae, Haptophyceae, Prasinophyceae, and Rhodophyceae. Cryobiology 52:152–156
Rippka R, Deruelles J, Waterbury JB, Herdman M, Stanier RY (1979) Generic assignments, strain histories and properties of pure cultures of Cyanobacteria. J Gen Microbiol 111:1–61
Romo S, Becares E (1992) Preservation of filamentous cyanobacteria cultures (Pseudanabaena galeata) Böcher and Geitlerinema amphibium (Ag. ex Gom.) Anagn. under low temperatures. J Microbiol Meth 16:85–89
Ruffing AM (2011) Engineered cyanobacteria: teaching an old bug new tricks. Bioeng Bugs 2(3):136–149
Sant'Anna CL, De Paiva Azevedo MT, Fiore MF, Lorenzi AS, Kastovsky J, Komarek J (2011) Subgeneric diversity of Brasilonema (Cyanobacteria, Scytonemataceae). Rev Bras Bot 34:51–62
SAEG (2007) Sistema de análises estatísticas e genéticas: versão 9.1. Viçosa: Fundação Arthur Bernardes
Silva PG, Ferrari SG, Silva HJ (2007) Preservation methods of Tolypothrix tenuis for use as a cyanobacterial fertilizer. J Appl Phycol 19:239–246
Simione FP (1998) Cryopreservation manual American type culture collection. In. Nalge Nunc International Corp, Rockville, pp 1-15
Smith D, Onion AHS (1983) The preservation and maintenance of living fungi. Veterinary dermatology, vol 4. Commonwealth Mycological Institute, Kew
Smith D, Ryan MJ, Day JG (2001) The UK National Culture Collection (UKNCC) biological resource: properties, maintenance and management. Bakeham Lane, Egham, Surrey
Sojka JE, Brisson-Kimmick SV, Carlson GP, Coppoc GL (1990) Dimethyl sulfoxide update-new applications and dosing methods. Proc Ann Conv Am Assoc Equine Pract 36:683–690
Tan CS (1997) Preservation of fungi. Cryptog Mycolog 18:157–163
Taylor R, Fletcher RL (1998) Cryopreservation of eukaryotic algae—a review of methodologies. J Appl Phycol 10:481–501
Watanabe A (1959) Some devices for preserving blue-green algae in viable state. J Gen Appl Microbiol 5:153–157
Wilde A, Dienst D (2011) Tools for genetic manipulation of cyanobacteria. Bioenergetic processes of cyanobacteria: from evolutionary singularity to ecological diversity. Springer, Dordrecht
Yu Z-W, Quinn P (1994) Dimethyl sulphoxide: a review of its applications in cell biology. Biosci Rep 14:259–281
Acknowledgments
We would like to thank Prof. Rosane Aguiar (in memoriam) for sharing knowledge over years of guidance and continuous support of this work. Discussions with Prof. Antônio Galvão Nascimento (Universidade Federal de Viçosa) were very important for the development of this work. Scholarships granted by the Foundation for Research Assistance of Minas Gerais (FAPEMIG to AA Esteves-Ferreira and R. Loterio) are gratefully acknowledged.
Conflict of interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Esteves-Ferreira, A.A., Corrêa, D.M., Carneiro, A.P.S. et al. Comparative evaluation of different preservation methods for cyanobacterial strains. J Appl Phycol 25, 919–929 (2013). https://doi.org/10.1007/s10811-012-9927-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10811-012-9927-9