
Abstract
Attention deficit/hyperactivity disorder (ADHD) symptoms are common in youth with autism spectrum disorders (ASD) and are frequently treated with stimulant medications. Twenty-seven children were randomized to different dose titration schedules, and ADHD symptoms, tolerability, and aberrant behaviors were assessed weekly during a 6-week trial with long-acting liquid methylphenidate (MPH). MPH at low to moderate doses was effective in reducing ADHD symptoms and was well tolerated in young children with ASD and ADHD. Future studies are needed to assess generalization and maintenance of efficacy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Attention deficit/hyperactivity disorder (ADHD) symptoms are reported in 22–85% of children with autism spectrum disorders (ASD) (Gadow et al. 2006; Goldstein and Schwebach 2004; Lee and Ousley 2006; Rao and Landa 2013). Stimulant medications are frequently utilized in youth with ASD and/or other neurodevelopmental disorders. For example, in a nationally representative study of adolescents ages 13–17 in special education, 33% of adolescents with ASD and ADHD were receiving stimulant medications (Frazier et al. 2011).
Notwithstanding the common clinical use of stimulants, mixed results have been reported from studies examining stimulants in youth with ASD and ADHD. While early case and retrospective studies of stimulant medications in ASD reported only modest benefit and showed worsening of disruptive and stereotyped behaviors (Campbell et al. 1972; Schmidt 1982; Volkmar et al. 1985), irritability and social withdrawal (Handen et al. 2000), and poor tolerability (Stigler et al. 2004), more recent studies reported more favorable effects (Pearson et al. 2013; Quintana et al. 1995; Santosh et al. 2006). Notably, most of these studies [with an exception of Pearson et al. (2013)] examined the effects of immediate release (IR) stimulants, and the efficacy and tolerability data for extended release (ER) formulations were largely derived from studies in typically developing youth with ADHD.
A recently developed extended release oral suspension preparation of MPH (Quillivant XR) has been shown to decrease ADHD symptoms in children with ADHD, with an optimal dose range of 20 to 60 mg (Robb et al. 2014). In addition to providing more consistent coverage compared to IR preparations, because it is in a liquid form, it can be precisely titrated and dosed and provides an option for children who have difficulty swallowing pills. In this preliminary study, we sought to evaluate the efficacy, safety, and tolerability of this preparation, given at various doses, on ADHD symptoms in children with comorbid ASD and ADHD. We hypothesized that there would be a linear dose response effect of MPH dose on ADHD symptoms. We also explored whether or not age had moderating effects, as previous studies have demonstrated lower stimulant response rates in young children (compared to older children) (Greenhill et al. 2006).
Methods
Participants
Children were recruited from an Autism Center, Child Psychiatry Clinic, and from a summer treatment program for children with social impairments and ASD and/or ADHD. All participating children received a physical exam and screening labs including complete blood count with differential, basic metabolic panel, urine toxicity screen, pregnancy test (for females of child-bearing potential) as well as a 12-lead electrocardiogram (ECG). Vital signs including height, weight, temperature, blood pressure, and pulse rate were obtained at baseline and reviewed at each follow-up visit.
To be eligible for the study, participants had to meet the following inclusion criteria: (a) ages between 5 and 17 years; (b) a clinical diagnosis of Autistic disorder or Asperger’s disorder by DSM-IV-TR (APA 2000) or ASD by DSM-5 (APA 2013); (c) a diagnosis of ADHD based upon the Kiddie Schedule for Affective Disorders and Schizophrenia for School-Age Children–Present and Lifetime (K-SADS-PL) (Kaufman et al. 1997); (d) Clinical Global Impressions—Severity for ADHD (CGI-S-ADHD) rating of 4 (moderate severity) or higher; (e) findings on physical exam, labs and ECG judged to be normal for age with pulse and blood pressure within 95% of age and gender mean; (f) a parent or legal guardian available for informed consent and assent for children with developmental age 7 years or older; and (g) at least one parent fluent in English.
Children were excluded if they: (a) had a history of seizure disorder (febrile seizures were non-exclusionary); (b) hypersensitivity to methylphenidate or other components of this formulation (judged by clinician); or (c) significant cardiac or other medical contraindications for stimulant medication. Concomitant psychotropic medications other than stimulants were permitted as long as the dose had been stable for at least 4 weeks prior to screening (Table 1). All procedures performed were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Study Design, Procedures, and Measures
This was a parallel group 6-week trial of liquid formulation of MPH ER. After screening, eligible participants were randomized to one of the three flexible dose titration schedules: Very Low Dose Group (starting dose of 5 mg/day with potential max dose of 10 mg/day), Low Dose Group (starting dose of 5 mg/day with potential max dose of 20 mg/day), or Moderate Dose Group (starting dose of 5 mg/day with potential max dose of 40 mg/day) (Table 2).
In cases of poor tolerability, the dose was reduced to the prior level. Blinded (to the treatment group) clinician-raters performed independent evaluations of ADHD symptoms and overall impairment at each visit. At study conclusion, the maximum dose tolerated in the Moderate Dose Group was 20 mg/day, with exception of two children who received 30 mg/day. Therefore, the Low and Moderate Dose Groups were combined and renamed as Medium Dose Group. The mean dose at week 6 in this group was 20.28 mg (range = 10–30 mg, 0.5 mg/kg). The medium dose group was compared with the Very Low Dose Group renamed as Low Dose Group, whose mean dose at week 6 was 9.72 mg (range = 7.5–10 mg, 0.29 mg/kg).
Compliance was assessed by parent/caregiver interview and dose diary; non-adherence was defined as any week where more than 25% of doses were missed on any measure.
Efficacy Measure
A clinician blinded to the dosing schedule and adverse effects completed the ADHD Rating Scale, Investigator Version (ADHD RS-INV) weekly to assess the frequency of each ADHD symptom. A total raw score change from baseline (screening/baseline visit) to the end of the treatment (week 6) was the primary efficacy measure. The Clinical Global Impressions—ADHD, Improvement scale (CGI-I) was also a key efficacy measure to assess global improvement (or worsening) of ADHD symptoms compared to baseline.
Secondary Measure
Each week parents completed the aberrant behavior checklist (ABC) (Aman et al. 1985) to assess treatment-related irritability, social withdrawal (lethargy), and stereotypy, and the hyperactivity, attention, learning problems (HALP) sleep questionnaire (Stein et al. 2001) to assess common sleep problems.
Exploratory Measure for Moderator Effects
We examined the effects of age on treatment response.
Tolerability and Safety Measures
Vital signs including weight, height, temperature, blood pressure and pulse rates, were obtained and reviewed at each visit. Response Impressions and Side Effects Checklist-Kids (RISC-K) is a 38-item parent rating scale developed by the authors which included an expanded symptom list from the original Stimulant Side Effect Questionnaire (Barkley et al. 1990). The 10 most frequently endorsed items rated “severe” (>7) were analyzed. Suicidality was assessed with the Columbia Suicide Severity Rating Scale (C-SSRS) (http://cssrs.columbia.edu/) at screening/baseline, week 3, and week 6.
Statistical Methods
Unless otherwise noted, all analyses were conducted in Stata 14.0 (StataCorp, College Station, Texas). A repeated measures mixed model was used to assess the effect of liquid MPH ER dose over 6 weeks on ADHD symptoms. A post-hoc test of trend over time was also conducted in each treatment group, as were tests of simple effects of treatment at each time point. We assessed moderating effect of age on ADHD outcomes by including interaction terms between age and treatment group in the models for ADHD outcomes.
Statistical significance of differences in frequency was assessed using Fisher’s exact test. Weekly recorded vitals (pulse, weight, systolic and diastolic blood pressure) of the two treatment groups were also summarized and compared using Wilcoxon rank-sum tests.
Results
Subject recruitment and flow are described in Consort diagram (Fig. 1). Twenty- seven children met inclusion criteria. At the conclusion of study, 9 were randomized to the new Low Dose Group and 18 were randomized to the new Medium Dose Group. The demographic and baseline characteristics of the intent-to-treat (ITT) population are shown in Table 3. The majority of the subjects were male (92.6%, n = 25). In addition to ASD and ADHD, 37.0% (n = 10) of participants met criteria for Oppositional Defiant Disorder (ODD).

The CONSORT diagram shows enrollment to group allocation. The medium dose group was compiled from the groups initially labeled low and moderate dose groups
Dose Group, Time, and Age Effects on ADHD RS-INV and CGI (Figs. 2, 3)

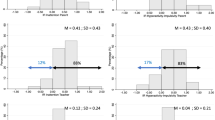
Adjusted predictions of treatment group and time effects on ADHD-RS-IV
ADHD symptoms declined for both groups over the course of the study (df = 6, X 2 = 130.30, p < 0.0001). There was a significant linear trend for declining ADHD RS-INV scores for both low and medium dose groups (p < 0.001 in both cases), without significant interaction between treatment groups (df = 6, X 2 = 10.75, p = 0.097). Treatment effects (Medium vs. Low Dose) on ADHD outcomes did not differ by age.
Improvement on CGI-I was more common for the Medium Dose Group (Fig. 3). By the end of trial (week 6), 33% of the Low Dose Group and 83% of the Medium Dose Group achieved a rating of “much improved” or “very much improved” on CGI-I. The effect of time (week) on CGI-I ratings was statistically significant for the Medium Dose Group (Medium Dose vs. Low Dose, z = 2.78, p = 0.005).

Adjusted predictions of the treatment group and time effects on CGI-I
Dose and Time Effects on ABC subscales and HALP Sleep Problems
There were no significant changes on ABC subscales between baseline and week 6 for the low dose group. However, Medium Dose Group displayed improvements on all five ABC subscales (Table 4). There were no significant group differences in frequencies of individual sleep problems at baseline, week 2, and week 6 on HALP sleep questionnaire, and only one child in the Medium Dose Group was rated as having a severe sleep problem at week 6. Excessive latency to sleep onset (>30 min) was the most common sleep problem, reported in 37% (n = 10) at baseline, 26% (n = 7) at week 2, and 26% (n = 7) week 6.
Safety and Tolerability by Treatment Group
There were no serious adverse events or suicidal behaviors reported. When comparing the Low Dose Group to the Medium Dose Group, there was a trend of increased pulse for the Medium Dose Group (p = 0.07), but there were no statistically significant or clinically significant changes for any of the vital signs. There were no dose-related increases in severe side effects assessed with the RISC-K over the course of the study. Interestingly, “rebound at end of day,” “aggression,” and “irritable” showed a tendency to decrease from baseline for the Medium Dose Group (Table 5).
Discussion
In this study, children with ASD and ADHD displayed linear reductions in ADHD symptoms regardless of the dosing group they were assigned to, and the dose response curves were similar to those of ADHD youth without ASD (Stein et al. 2003). However, clinically significant improvements in ADHD symptoms were much more common in youth in the medium dose group, which was also associated with improvements in all five ABC subscales, including irritability, lethargy, stereotyped behaviors, hyperactivity, and inappropriate speech.
Tolerability and compliance were better than expected from previous studies, likely because investigators were allowed to use their clinical judgement to adjust the dose proactively rather than pushing the dose to the maximum dose set for the assigned group. While one participant discontinued treatment participation prematurely, it was due to issues related to scheduling conflicts, not due to tolerability. No dose-dependent increases in insomnia or other adverse events were observed within the relatively low dose ranges and gradual titration strategy utilized in this study.
Unlike previous studies that demonstrated lower stimulant response rates in young children (compared to older children) (Greenhill et al. 2006), we did not find age to be a significant moderator of treatment response in this study. This may be due to our minimum age limit set at 5 years [older than those in the Preschool ADHD Treatment Study (PATS)] and relatively restricted age range.
Our findings should be viewed in light of several methodological limitations. First, as this was a pilot study with a modest sample size, we did not control for multiple comparisons. Second, we did not have a placebo arm to control for expectancy effects and parents were aware of titration changes. Third, although sleep problems were modest and not related to the liquid MPH ER dose or time, it should be noted that 37% of the children were receiving a sleep medication (e.g., melatonin, alpha-2 agonist at baseline) and were allowed to continue during the course of the study. Fourth, this was a single site study of predominantly boys and relatively homogenous in terms of ethnicity and social economic status; therefore, findings may not be generalizable to other samples. Fifth, since many of the study participants enrolled during the summer, we were not able to utilize teacher ratings to assess outcomes on classroom symptoms and functioning. Similarly, we did not formally perform neuropsychological testing (e.g., cognitive testing) to better characterize the sample. In addition, we included participants with a clinical diagnosis of ASD based on either DSM-IV-TR or DSM-5. It is unlikely, however, that subtle differences in diagnostic criteria or procedures would greatly influence the robust findings.
In summary, liquid MPH ER appeared to be effective at low- to moderate dose ranges (10–30 mg daily) in reducing ADHD symptoms and was well tolerated in our study participants. Clinically significant improvements in ADHD symptoms were noted more frequently in children in the Medium Dose Group (mean dose = 20.28 mg/day) with adequate tolerance to stimulant side effects. Future studies are needed to replicate findings and examine moderators of response with a larger sample of youth with ASD and ADHD to assess generalization and to examine the long-term impact on impairment and functioning.
References
Aman, M. G., Singh, N. N., Stewart, A. W., & Field, C. J. (1985). The aberrant behavior checklist: A behavior rating scale for the assessment of treatment effects. American Journal of Mental Deficiency, 89, 485–491.
APA. (2000). Diagnostic and statistical manual of mental disorders, text revision (DSM-IV-TR), (4th ed.). Washington, DC: American Psychiatric Publishing.
APA. (2013). Diagnostic and statistical manual of mental disorders, Fifth Edition (DSM-5). Washington, DC: American Psychiatric Publishing.
Barkley, R. A., McMurray, M. B., Edelbrock, C. S., & Robbins, K. (1990). Side effects of methylphenidate in children with attention deficit hyperactivity disorder: A systemic, placebo-controlled evaluation. Pediatrics, 86, 184–192.
Campbell, M., Fish, B., David, R., Shapiro, T., Collins, P., & Koh, C. (1972). Response to triiodothyronine and dextroamphetamine: A study of preschool schizophrenic children. Journal of Autism and Developmental Disorders, 2(4), 343–358.
Frazier, T. W., Shattuck, P. T., Narendorf, S. C., Cooper, B. P., Wagner, M., & Spitznagel, E. L. (2011). Prevalence and correlates of psychotropic medication use in adolescents with an autism spectrum disorder with and without caregiver-reported attention-deficit/hyperactivity disorder. Journal of Child and Adolescent Psychopharmacology, 21, 571–579.
Gadow, K. D., DeVincent, C. J., & Pomeroy, J. (2006). ADHD symptom subtypes in children with pervasive developmental disorder. Journal of Autism and Developmental Disorders, 36, 271–283.
Goldstein, S., & Schwebach, A. J. (2004). The comorbidity of pervasive developmental disorder and attention deficit hyperactivity disorder: Results of a retrospective chart review. Journal of Autism and Developmental Disorders, 34, 329–339.
Greenhill, L., Kollins, S., Abikoff, H., McCracken, J., Riddle, M., Swanson, J., et al. (2006). Efficacy and safety of immediate-release methylphenidate treatment for preschoolers with ADHD. Journal of the American Academy of Child and Adolescent Psychiatry, 45, 1284–1293.
Handen, B. L., Johnson, C. R., & Lubetsky, M. (2000). Efficacy of methylphenidate among children with autism and symptoms of attention-deficit hyperactivity disorder. Journal of Autism and Developmental Disorders, 30, 245–255. doi:10.1023/A:1005548619694.
Kaufman, J., Birmaher, B., Brent, D., Rao, U., Flynn, C., Moreci, P., et al. (1997). Schedule for affective disorders and schizophrenia for school-age children-present and lifetime version (K-SADS-PL): Initial reliability and validity data. Journal of the American Academy of Child and Adolescent Psychiatry, 36, 980–988.
Lee, D. O., & Ousley, O. Y. (2006). Attention-deficit hyperactivity disorder symptoms in a clinic sample of children and adolescents with pervasive developmental disorders. Journal of Child and Adolescent Psychopharmacology, 16, 737–746.
Pearson, D. A., Santos, C. W., Aman, M. G., Arnold, L. E., Casat, C. D., Mansour, R., … Cleveland, L. A. (2013). Effects of extended release methylphenidate treatment on ratings of attention-deficit/hyperactivity disorder (ADHD) and associated behavior in children with autism spectrum disorders and ADHD symptoms. Journal of Child and Adolescent Psychopharmacology, 23(5), 337–351. doi:10.1089/cap.2012.0096.
Quintana, H., Birmaher, B., Stedge, D., Lennon, S., Freed, J., Bridge, J., & Greenhill, L. (1995). Use of methylphenidate in the treatment of children with autistic disorder. Journal of Autism and Developmental Disorders, 25(3), 283–294.
Rao, P. A., Landa, R. J. (2013). Association between severity of behavioral phenotype and comorbid attention deficit hyperactivity disorder symptoms in children with autism spectrum disorders. Autism, 18, 272–280.
Robb, A. S., Findling, R .L., Childress, A. C., Berry, S. A., Belden, H. W., Wigal, S. B. (2014). Efficacy, safety, and tolerability of a novel methylphenidate extended-release oral suspension (MEROS) in ADHD. Journal of Attention Disorders. doi:10.1177/1087054714533191.
Santosh, P. J., Baird, G., Pityaratstian, N., Tavare, E., Gringas, P. (2006). Impact of comorbid autism spectrum disorders on stimulant response in children with attention deficit hyperactivity disorder: A retrospective and prospective effectiveness study. Child: Care, Health, and Development, 32, 575–583. doi:10.1111/j.1365-2214.2006.00631.x.
Stein, M. A., Mendelsohn, J., Obermeyer, W. H., Amromin, J., & Benca, R. (2001). Sleep and behavior problems in school-aged children. Pediatrics, 107, E60.
Stein, M. A., Sarampote, C. S., Waldman, I. D., Robb, A. S., Conlon, C., Pearl, P. L., et al. (2003). A dose-response study of OROS methylphenidate in children with attention-deficit/hyperactivity disorder. Pediatrics, 112, e404.
Schmidt, K. (1982). The effect of stimulant medication in childhood-onset pervasive developmental disorder—a case report. Journal of Developmental & Behavioral Pediatrics, 3(4), 244–246.
Stigler, K. A., Desmond, L. A., Posey, D. J., Wiegand, R. E., & McDougle, C. J. (2004). A naturalistic retrospective analysis of psychostimulants in pervasive developmental disorders. Journal of Child and Adolescent Psychopharmacology, 14, 49–56.
Volkmar, F. R., Hoder, E. L., Cohen, D. J. (1985). Inappropriate uses of stimulant medications. Clinical Pediatrics, 24, 127–130.
Author Contributions
All authors were involved in study conduct and contributed to the manuscript.
Funding
This study was an investigator initiated study funded by Pfizer Inc© (WI185890). Preliminary results of this study were presented at the American Professional Society of ADHD and Related Disorders’ annual meeting at Washington D.C. in January 2016 and the Pediatric Academic Societies’ annual meeting at Baltimore, Maryland in May 2016.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Mark A. Stein, PhD, receives research support from Akili Interactive Labs, National Institute of Mental Health, Pfizer Inc, and Shire. Mark A. Stein, PhD, is also a consultant for Akili Interactive Labs, Alcobra Pharma, Arbor Pharmaceuticals, Ironshore Pharmaceuticals and Development, Lundbeck, Shire, and Sunovion Pharmaceuticals Inc. Drs. Kim, French, Strickland, and Ms. Miller and Shonka do not have any conflicts of interest.
Rights and permissions
About this article

Cite this article
Kim, SJ., Shonka, S., French, W.P. et al. Dose-Response Effects of Long-Acting Liquid Methylphenidate in Children with Attention Deficit/Hyperactivity Disorder (ADHD) and Autism Spectrum Disorder (ASD): A Pilot Study. J Autism Dev Disord 47, 2307–2313 (2017). https://doi.org/10.1007/s10803-017-3125-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10803-017-3125-1