
Abstract
In order to provide an alternative approach for understanding the biology and genetics of autism, we performed statistical analysis of gene expression profiles of lymphoblastoid cell lines derived from children with autism and their families. The goal was to assess the feasibility of using this model in identifying autism-associated genes. Replicate microarray experiments demonstrated that expression data from the cell lines were consistent and highly reproducible. Further analyses identified differentially expressed genes between cell lines derived from children with autism and those derived from their normally developing siblings. These genes were then used to identify biochemical pathways potentially involved in autism. This study suggests that lymphoblastoid cell lines may be a viable tool for identifying genes associated with autism.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Autism is a complex neurodevelopmental disorder characterized by deficits in communication, social reciprocity, and a pattern of repetitive or stereotypic behaviors and interests (American Psychiatric Association, 1994; World Health Organization, 1993). Autism presents significant diagnostic and therapeutic challenges due to its clinical heterogeneity, lack of diagnostic markers, inadequate understanding of the underlying pathophysiology, and its complex genetic architecture (Bailey et al., 1995; Lord et al., 2000).
Until recently, identification of genes associated with autism has been based on linkage analysis using microsatellite markers to identify potential susceptibility regions in the genome (Buxbaum et al., 2001; Liu et al., 2001; Philippe et al., 2002; Risch et al., 1999). The systematic search for linked loci has not yielded definitively associated genes, in part due to the heterogeneity of the clinical spectrum of the disease, and it has become quite clear that autism is a complex disease resulting from collective actions of many genes (Buxbaum et al., 2001; Pickles et al., 1995; Purcell et al., 2001; Risch et al., 1999). Consequently, genetic dissection of autism needs to be carried out in a global context.
Genomics, the comprehensive study of genes and their functions, offers a unique approach to the genetics of autism because it lends the ability to study the disorder in a systematically global fashion. Gene expression microarray technology has been one of the foundations of genomics, and it provides a tool that can capture the expression of essentially all genes simultaneously, yielding whole-genome molecular profiles of cells (Geschwind & Gregg, MIT Press, 2002). Oligonucleotide microarrays consist of thousands of individual DNA sequences (25–60 bp fragments) representing individual genes placed onto a silicon wafer to form a microarray chip. RNA from cells (i.e. cultured cells, tissue, or blood) is processed, labeled, and hybridized onto the microarray chip. Because the RNA sample is labeled with a fluorescent tag, the amount of fluorescence at each oligonucleotide corresponds with the abundance of the specific mRNA (gene) in the sample. Such analysis yields the gene expression “fingerprints” of all the RNA transcripts (transcriptome), which reflect the state of the cell or tissue at the time of RNA harvest. Microarrays, therefore, offer a unique and alternative approach for identifying and determining functions of genes associated with complex neurodevelopmental diseases (reviewed in (Luo & Geschwind, 2001)). Further, data from gene expression microarrays can also be used to map complex quantitative traits (Cheung et al., 2003; Schadt et al., 2003), another significant contribution to the study of a complex spectrum disorder such as autism.
To date, the application of microarray technology to autism research has been limited. Purcell et al. (Purcell et al., 2001) have used cDNA microarrays to study gene expression in postmortem brain tissues from individuals with autism. However, the number of postmortem brain tissues from individuals with autism is limited, which potentially erodes this avenue of analysis. An alternative that has emerged is the field of blood genomics, where peripheral lymphocytes are used to represent the transcriptome. Blood genomics is beginning to be applied to the study of central nervous system (CNS) disorders; studies have demonstrated that genes expressed in the peripheral blood can classify CNS disorders and may be able to identify and predict the genes and pathobiologies involved in these disorders (Lu et al., 2003; Tang et al., 2004; Tang, Nee, Lu, & Sharp, 2003).
The objective of our pilot study was to provide proof of principle experiments showing that cell lines derived from peripheral lymphocytes—from children with autism and from age-matched normally developing siblings—can be used to dissect autism on a whole-genome basis. In order to validate this approach, we identified genes that are differentially expressed between the cell lines derived from children with autism and the cell lines derived from age-matched normally developing siblings, and we clustered the two groups into their respective phenotypes. In addition, we investigated the reproducibility of the lymphoblastoid cell line data used to identify differentially expressed genes. These experiments suggest that gene expression profiling of cell lines may provide a valuable link to identifying genes associated with autism.
Materials and Methods
Cell Lines
The cell lines used in this study were human EBV-transformed lymphoblastoid cell lines derived from blood samples from children with autism and from age-matched normally developing siblings. The cell lines were obtained from the Autism Genetic Resource Exchange (AGRE), an internationally recognized central repository and family registry for autism, created by Cure Autism Now (CAN) and the Human Biological Data Interchange for autism studies. Family recruitment and diagnostic assessments were based on the procedures established by CAN and are fully described by (Geschwind et al., 2001). A complete description of each sample used in this study is available through AGRE at http://www.agre.org.
The selection criteria for cell lines used in this study required that one of at least two siblings be clinically diagnosed with autism using clinical diagnostic procedures as described by Lord et al. (Lord et al., 1997; Lord, Rutter & Le Couteur, 1994). The cell lines used in this study included HI-0128 (AU002905), HI-0158 (AU010904), and HI-0489 (AU014505), which are derived from blood samples drawn from children with autism. Cell lines HI-0130, HI-0157, and HI-0487 were derived from blood samples of age-matched normally developing siblings and were used as controls in the analysis. In particular, the cell line HI-0128 was derived from a female with autism (verbal with regressive autism), while HI-0130 was derived from her normally developing sister. In addition, this family has a son with autism that was not used in this study. The sibling pair HI-0157 and HI-0158 was from a set of fraternal triplets. The HI-0158 cell line was derived from a male with autism who is verbal, while HI-0157 was derived from his normally developing brother. The sibling pair HI-0487 and HI-0489 was also from a set of fraternal triplets. The cell line HI-0489 was derived from a non-verbal boy with regressive autism, while HI-0487 was derived from his normally developing fraternal brother. Table 1 summarizes the samples and provides information regarding the scores used to classify the patients as autistic.
Cell Culture
The cell lines were grown in suspension in RPMI-1640, 15% fetal bovine serum (FBS), 50 units/ml penicillin, 50 μg/μl streptomycin, in 5% CO2-humidified tissue culture incubator. Cells were harvested during the logarithmic growth phase, taking particular care to minimize experimental variability that might lead to differences in gene expression.
To evaluate the reproducibility of data, we replicated cell line HI-0489 four times by culturing it simultaneously in four separate flasks and performing gene expression profiling on each replicate sample. The use of replicate material from the same cell line eliminated genetic heterogeneity and variability, thereby providing material for studying reproducibility and any systematic variation.
Gene Expression Analysis
Total RNA was isolated immediately upon cell harvest, as described by the TRIzol Reagent (Invitrogen, Carlsbad, CA) RNA isolation procedure, and further purified using the RNeasy kit (Qiagen, Chatsworth, CA). RNA quality was assessed using the Agilent 2100 Bioanalyzer (Agilent Technologies Inc.). The RNA samples were then processed and hybridized to Human Genome GeneChip U95Av2 (HG-U95Av2, Affymetrix Inc., Santa Clara, CA) according to the manufacturer’s protocol and as previously described (Rea, Gregg, Qin, Phillips, & Rice, 2003). The U95Av2 GeneChip represents 12,600 well-annotated genes and expressed sequence tags (ESTs) from UniGene Build 95.
Microarray Data Processing and Analysis
Gene expression values were calculated from the scanned image obtained from the U95Av2 Human Genome GeneChip using Microarray Analysis Suite (MAS) 5.0 from Affymetrix, giving a single average scaled difference value across 16–20 probe pairs as well as a detection reliability score [Absent (A), Present (P), or Marginal (M), based on the variability of hybridization within each probe set], detection P-value, and comparison P-value. Expression values were adjusted for background and noise level (experimental and other unknown sources of variation), globally normalized, and scaled to a mean value of 125 to allow for interchip comparisons using MAS 5.0.
Reproducibility
Preliminary analysis was conducted using SAS (SAS, Inc., Cary, NC) statistical packages on the four replicates of the same cell lines to assess the reproducibility of data. We then performed pair-wise comparisons using the Pearson correlation coefficient, and used analysis of variance (ANOVA) for simultaneous comparison of the four replicates to determine if the difference in gene expression values among the four replicates was significant. We assessed the measurement error using the coefficient of variation for each gene using expression values of the four replicates. In addition, we assessed reproducibility at the probe level by ranking genes based on detection P-values obtained from each replicate. To insure that the correlation was not obtained simply by chance, we performed a t-test on the identified genes using replicate data, treating replicates 1 and 2 as Group 1, and replicates 3 and 4 as Group 2 using the criteria for identifying genes. Furthermore, we calculated the Pearson correlation for identified genes using mean expression values of replicates for individual genes from the two replicate groups, and we compared the resulting correlation to the correlation obtained between mean expression values from children with autism and their normally developing siblings.
Differential Gene Expression
In order to identify differentially expressed genes between cell lines derived from children with autism and cell lines derived from normally developing siblings, we filtered MAS 5.0 generated expression values using the following conditions: a particular transcript was considered not expressed if it was marked absent across the six arrays (i.e., not detected) and was not considered for further analysis. In addition, we filtered out the spiked-in control genes. This filtering left 6,563 (56%) of the genes for further analysis.
The gene expression values were log transformed (log base 2) so that the expression of genes emulated a normally distributed dataset. To test the hypothesis that the gene expression in cell lines from children with autism may be different from expression in cell lines from normally developing siblings, we performed a t-test on the log transformed data using the bootstrap method (Kerr & Churchill, 2001) to determine the significance level of the differences in expression between the two groups. The fold difference was determined by the mean-difference of the log-transformed data between the groups. Genes were ranked on t-test scores, P-values (P ≤ 0.05), and fold changes.
Selection of the differentially expressed genes was based on the following criteria: a particular transcript was considered significantly differentially expressed between the two groups if the P-value was less than 0.05, its expression was greater than 60 intensity units (greater than the average background), and had a fold change (θ) of θ ≥ 1.30 as measured based on the actual expression values. These parameters permit identification of differentially expressed genes with biological meaning while limiting the number of false positives and ensuring reliability of detection. We computed the Euclidean distances between genes and between cell lines, and used hierarchical clustering via a method of complete linkage (Herrero et al., 2003) to cluster genes into autism and normal phenotypic classes.
Statistical computing was done using MATLAB (MathWorks, Inc., Natick, MA) and SAS statistical packages.
Pathway Identification
In an effort to understand the potential relationships between the differentially expressed genes, we utilized the PathwayAssist software package (Stratagene, La Jolla, CA), which is a visualization tool that represents functional relationships between genes in a graphical front-end. The software relies upon a collection of databases, including: Kyoto Encyclopedia of Genes and Genomes (KEGG), Database of Interacting Proteins (DIP), and Biomolecular Interaction Network Database (BIND), in addition to the proprietary database, ResNet.
ResNet is compiled quarterly using proprietary Natural Language Processing algorithms to extract information from the entire PubMed library. Current versions of the ResNet database contain over 100,000 events of regulation, interaction, and modification of gene products. PathwayAssist enables the user to query genes of interest and generate potential pathways of interaction based on a wealth of published data. User-directed pathways can be constructed through pathway-generating actions, querying the ResNet database for a host of interactions among selected genes, including direct interactions and intermediary interactions. Synopses of the software-generated interactions can be reviewed by the user to enable user-directed curation. All visualizations generated by PathwayAssist can be readily exported into standard image formats for easy integration into presentation or publication formats.
In order to identify inter-relationships between the differentially expressed genes, the list of Affymetix probe IDs passing the selection criteria described in the Microarray Data Processing and Analysis section above was entered into the PathwayAssist program. The probes recognized by the software’s database were mapped to proteins which were made available for querying and analysis. The ResNet database was then queried using the “Shortest Path between Probes” function to generate a network of interactions. In an effort to reduce the number of irrelevant interactions, those proteins with excessive numbers of connections were excluded from the above step.
All proteins in the resulting workspace, including those that were excluded for having excessive numbers of connections, were selected and queried for “Direct Interactions”, to elucidate any potential connections that may have been overlooked. The entire network of nodes generated by the program was manually inspected and curated for dubious or redundant interactions. All “Unlinked Nodes” were removed from the network. Those proteins in the final pathway that were derived from the list of differentially expressed genes were overlaid in color to represent changes in gene expression.
Results
Our goal was to evaluate reproducibility of gene expression data, to identify a set of differentially expressed genes between the cell lines derived from the children with autism and their normally developing siblings, and to determine if identified differentially expressed genes could distinguish the two groups into their clinical phenotypes.
Reproducibility
As a first step toward determining the sensitivity at which we could identify differentially expressed genes, we evaluated reproducibility of gene expression data using expression values obtained from four replicates. Pair-wise comparison using the Pearson correlation showed that the expression values were consistent and highly reproducible. The average pair-wise Pearson correlation was 0.99, with a range of 0.98–0.99. Simultaneous comparison of expression values from all four replicates using ANOVA showed no significant difference (P > 0.05) among the four replicates. These data suggest that GeneChip microarray gene expression data from cell lines is consistent and highly reproducible, and that genes identified as differentially expressed represent true variation rather than experimental effect.
Differentially Expressed Genes
Using t-test analysis on the cell lines derived from children with autism and the cell lines derived from their normally developing siblings, we identified 48 significantly (P ≤ 0.05) differentially expressed up- or down-regulated genes between cell lines derived from children with autism and their normally developing siblings. Table 2 shows a list of these 48 genes along with their P-value and fold change. The identified genes had fold changes ranging from −6.32 for IGHG3 to 3.92 for IGL3. We identified genes with diverse molecular functions, including genes involved in signal transduction, metabolism, cell growth and maintenance, genes involved in response to external stimuli, and genes involved in development. Genes involved in signal transduction and regulation of signaling pathways included PAK1, INPP5D, FNTA, C6orf4, PDE8A, GNB2, LANCL1, ARCHGAP4, GNA15, SIT, ARHGEF1 and PNOC. Also identified in this functional group were genes involved in cell signaling and cell adhesion, ITGB7, FLOT2, ITGB1, and the cell adhesion molecule antigen, CD44, suggesting that cell signaling may be an important aspect of the pathogenesis of autism.
Among the identified genes involved in metabolism and regulating metabolic pathways were PAK1, INPP5, IGHMB2, GBE1, BTN2A1, PRCP, STARD3, PDE8A, GNA15 and JUNB. The other prominent group among the identified genes were genes involved in cell growth and maintenance, which included a gene involved in biogenesis (ARHGAP4) and genes involved in cell proliferation (IGHMBP2 and ARHGEF1). Other genes within this functional group included STARD3, ARHGAP4, and KCNN4, as well as genes involved in development, which included ITGB7, FLOT2, and TPM4. In addition, we identified genes that respond to exogenous stimuli (ITGB1, CD74, IGHG3, KCNN4, and IGL3) and a gene that responds to endogenous stimuli (IGHMBP2), indicating that the immune system may be playing an important role in the pathogenesis of autism.
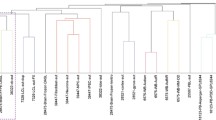
To evaluate whether the dichotomy of the two groups could be corroborated using an alternate approach, the data from 48 significant differentially expressed genes were analyzed using hierarchical clustering. Figure 1 shows a dendrogram of two-way hierarchical clustering of genes and cell lines. The colors red and green indicate up-regulation and down-regulation, respectively. Our subset of genes generated two distinct types of gene expression profiles that could potentially be used as phenotype predictors of future analyses of cell lines derived from children with autism and cell lines derived from their age-matched normally developing siblings. It must be stated that this initial analysis was made with three subjects in each class and therefore larger studies will be needed to verify these findings and approach.

Two-way hierarchical clustering distinguishing gene expression in cell lines derived from children with autism from expression in cell lines derived from normally developing siblings. Sample n = 6; gene n = 48. The red and green colors indicate up and down regulation, respectively. HI-0128, HI-0158, HI-0489 are cell lines derived from children with autism, HI-0130, HI-0157, HI-0487 are cell lines derived from their normally developing siblings
As a step towards understanding the relationship between reproducibility and differential expression and testing the reliability of the results of our differential gene identification, we performed further tests on the identified genes using the replicate and experimental data sets as described in the data analysis section. Figure 2 shows linear scatter plots of mean expression values for the 48 identified genes for the 2 data sets as determined by the t-test and correlation. The two panels contrast the degree of correlation between the replicates with the degree of correlation between the autistic and non-autistic cell lines. There were no significant differences in gene expression among the replicates—with a correlation of r 2 = 0.994 (Fig. 2A) and a coefficient of variation of 0.0007. There were significant (P ≤ 0.05) differences in gene expression between cell lines derived from children with autism and cell lines from their normally developing siblings—with a correlation of r 2 = 0.801 (Fig. 2B) and a coefficient of variation of 0.8. These results suggest that, in this pilot study, the genes identified as important in distinguishing the autistic samples were differentially expressed and not identified by chance.

Linear scatter plot of mean expression values in log scale (log base 2) for technical replicates and biological replicates. (A) is a plot of mean expression values for replicate Group1 (using the mean of replicate 1 and replicate 2) against Group 2 (using the mean of replicate 3 and replicate 4) of the same cell line (HI-0489). (B) is a plot of mean expression values from children with autism (Group 1; n = 3) against values from normal developing siblings (Group 2; n = 3). r 2 indicates the correlation and red color indicates differential expression of each gene (gene n = 48)
Pathway Identification
With gene expression profiling, large lists of differentially expressed genes are generated, and the relationships between these genes can be difficult to visualize and interpret. PathwayAssist was utilized in order to identify potential pathways that may be involved in autism. PathwayAssist was queried with the list of 48 differentially expressed genes; it recognized 43 genes, which it used to generate networks of molecular interactions. All of the genes—except CD44, CD74, JUNB, and SDBCAG84—were used by the software to query multiple databases and generate a network of connections based on the shortest path between molecules. CD44, CD74, JUNB, and SDBCAG84 have been extensively reported to be associated with cancer and not currently with autism—as a result, there is abundant literature on these genes that creates a highly complex and overwhelming set of connections that obscure the connections between the majority of the genes in the list. Inclusion of these four genes in the query produced a pathway that contained 39 nodes and 284 referenced interactions. For this reason, these four genes were removed from the initial analysis.
Building on the shortest path network generated above, we re-integrated the four excluded proteins into the pathway by querying for direct interactions between all of the molecules. After removing redundant interactions and unlinked nodes, we resolved a molecular network which included 15 of the differentially expressed proteins, as well as interactions with 2 small molecules (Fig. 3).

PathwayAssist was queried using the list of 48 differentially expressed genes; it recognized 43 genes, which it used to generate a network of molecular interactions. The shapes of the nodes represent different structures and functions, as indicated by the legend. Relationships between the nodes are indicated by various types of connections (shown in legend), with ± indicating any positive or negative effect. Red indicates genes that are up-regulated in the cell lines derived from the group of children with autism, while green indicates genes that are down-regulated
Discussion
The use of peripheral blood in the study of autism has obvious benefits in that samples can be collected easily and non-invasively. In contrast, the use of postmortem brain (Purcell et al., 2001), where the pathobiology of autism is likely occurring, has merits—however, it is often not possible to obtain CNS tissue in the rapid fashion which is required for high fidelity RNA. Therefore, we believe the alternative approach of using peripheral lymphocytes has strong merits. This new paradigm of “blood genomics” in CNS disorders has already been demonstrated to be feasible. Gene expression profiles obtained from blood mRNA of mice have been used to identify neuronal injury (Tang et al., 2003). In addition, using gene expression of RNA from blood, a pediatric epilepsy group was differentiated from healthy volunteers, chronic daily headache pediatric patients, migraine pediatric patients, and adult patients with bipolar disorder or schizophrenia (Tang et al., 2004). These studies demonstrate that variations in the state of the CNS are reflected in the mRNA of the peripheral blood and are detectable by gene expression microarray.
As we postulate that the pathology of autism has a strong genetic component, it is tenable that cell lines derived from peripheral blood will carry a genetic signature that reflects the genetic aberrations associated with autism. This approach has already been applied to bipolar disorder, where XBP1 was identified as a genetic risk factor using gene expression profiling of lymphoblastoid cell lines from two sets of discordant twins (Kakiuchi et al., 2003). In our proof-of-principle study, we demonstrated that lymphoblastoid cell lines derived from the peripheral blood of children with autism can potentially be utilized to identify molecules that are associated with the disorder.
Although the initial dataset from our experiments is small, it is compelling that we were able to identify a set of genes that could then be used to distinguish the children with autism from the normally developing children. In our experience, the set of genes identified as differentially expressed via one method (in this case, through filtering criteria, t-test, and significance analysis) does not always segregate samples into the appropriate classification when used as the gene list for hierarchical clustering. We therefore used this approach as a test of the significance of these genes and their role in autism.
In addition by using replicate cell lines, our data show that the gene expression profiles are highly reproducible. Our average Pearson correlation of 0.99 (range of 0.98–0.99) obtained from pair-wise analysis of four replicates of the same cell line is consistent with correlations of 0.98–0.99 reported by Bakay et al. (Bakay et al., 2002) and Unger et al. (Unger et al., 2001) using the same microarray platform. These data suggest that the gene expression changes that we have identified are reproducible.
In the set of the genes that our study identified as differentially expressed in autism, we did not directly identify any genes that have been previously associated with autism. However, by applying pathway analysis methods to our gene list, we were able to detect known relationships between these genes and biological processes. As one example, we identified norepinephrine as a small molecule interacting with our set of differentially expressed genes. The metabolism of norepinephrine from dopamine is catalyzed by serum dopamine beta hydroxylase (DbetaH), and studies have found that low serum maternal DbetaH and genotypic susceptibility of this gene in the fetus is associated with autism (Robinson, Schutz, Macciardi, White, & Holden, 2001). It has been shown that abnormalities in serotonin (5-hydroxytryptamine or 5-HT), a related neurotransmitter, have been identified in roughly one-third of subjects with autism spectrum disorder and in first-degree relatives (Anderson et al., 1987). Linkage studies have found that the serotonin receptor is linked with autism (Yonan et al., 2003). In addition, it has been shown that children with autism and their first-degree relatives have elevated serotonin levels (Gingrich & Hen, 2001). In addition, clomipramine, a serotonin and norepinephrine reuptake inhibitor, has shown efficacy for certain symptoms of autism (anger, hyperactivity and repetitive behavior) in children, adolescents, and young adults (Gordon, State, Nelson, Hamburger, & Rapoport, 1993; Luiselli, Blew, Keane, Thibadeau, & Holzman, 2000). Other selective serotonin reuptake inhibitors (SSRIs) have shown promising results in ameliorating symptoms in children with autism (Posey, Litwiller, Koburn, & McDougle, 1999; Posey & McDougle, 2000).
Another finding potentially relevant to autism is the identification of several genes related to immune response. These genes have not been previously associated with autism, but their detection in our microarray experiments may tie in with the long-standing hypothesis of suppression or dysregulation of the immune system in individuals with autism (Warren et al., 1986). It is not clear from this study or from previous studies how the immune system is genetically regulated or dysregulated in individuals with autism, so these findings may provide an important starting point for dissecting the pathways involved in immune response and autism.
Although the genes identified in our study do not have roles in autism that have been clearly defined to date, they may be indicative of a portion of the intricate genetic network involved in autism. In addition, the identification of norepinephrine as a small molecule interacting with our set of differentially expressed genes suggests that we may be able to gain insight into the complex mechanisms underlying the patterns of neurotransmitter levels that are already associated with autism. Taken together, these data underscore the potential of this approach and that by applying unique analysis strategies one can identify cellular processes and pathways. To our knowledge this is the first description of gene expression microarray data in autism analyzed with pathway analysis software. Lastly, aside from the most obvious application, the identification of directly related genes, gene expression studies can also be used as a set of focused, hypothesis-generating experiments for the identification of relevant interacting processes and pathways.
As stated before, this study was performed as a set of proof-of-principle experiments, and the present results are based on a small set of cell lines. While these experiments produced some potentially promising results and a list of genes that may be—albeit indirectly—related to autism, a clear molecular portrait of autism can only be gleaned through further studies using larger sample sizes. Future studies will require larger sample sizes in order to allow reliable identification and characterization of all the differentially expressed genes involved in autism, full molecular classification of the subtypes of autism, and prediction of the efficacy of pharmacological intervention. Zien et al. (Zien, Aigner, Zimmer, & Lengauer, 2001) and recently, Mukherjee et al. (Mukherjee et al., 2003) have shown that a minimum sample size of 25 in each group will give statistical confidence in classifying related diseases or sub-phenotypes, while a sample size of 10 is needed to identify different groups of unrelated tissue or disease types. Therefore, the present study will need to be expanded using more samples to identify the set of genes that consistently accounts for variation in autism phenotypes and provides discrimination that allows classification and prediction of many subtypes of autism. These subsequent experiments are very feasible, as the field of autism research has generated rich resources of publicly available cell lines from children with autism and their families (Geschwind et al., 2001).
In conclusion, this pilot study demonstrates that there is the potential for the identification of genes that are differentially expressed between cell lines derived from children with autism and cell lines derived from their normally developing siblings If such genes could identified and verified in larger datasets, they could be used as molecular signatures for diagnosing autism, predicting subtypes of autism, or for the identification of therapeutic targets. This could provide the ability for families to begin early behavioral or pharmacological intervention for children likely to be at risk for autism.
References
American Psychiatric Association (1994). Diagnostic and statistical manual of mental disorders (4th ed.). Washington, DC.
Anderson, G. M., Freedman, D. X., Cohen, D. J., Volkmar, F. R., Hoder, E. L., McPhedran, P., Minderaa, R. B., Hansen, C. R., & Young, J. G. (1987). Whole blood serotonin in autistic and normal subjects. Journal of Child Psychology and Psychiatry, 28, 885–900.
Bailey, A., Le Couteur, A., Gottesman, I., Bolton, P., Simonoff, E., Yuzda, E., & Rutter, M. (1995). Autism as a strongly genetic disorder: Evidence from a British twin study. Psychological Medicine, 25, 63–77.
Bakay, M., Chen, Y. W., Borup, R., Zhao, P., Nagaraju, K., & Hoffman, E. P. (2002). Sources of variability and effect of experimental approach on expression profiling data interpretation. BMC Bioinformatics, 3, 4.
Buxbaum, J. D., Silverman, J. M., Smith, C. J., Kilifarski, M., Reichert, J., Hollander, E., Lawlor, B. A., Fitzgerald, M., Greenberg, D. A., & Davis, K. L. (2001). Evidence for a susceptibility gene for autism on chromosome 2 and for genetic heterogeneity. American Journal of Human Genetics, 68, 1514–1520.
Cheung, V. G., Conlin, L. K., Weber, T. M., Arcaro, M., Jen, K. Y., Morley, M., & Spielman, R. S. (2003). Natural variation in human gene expression assessed in lymphoblastoid cells. Nature Genetics, 33, 422–425.
Geschwind, D. H., & Gregg, J. P. (2002). Microarrays for the Neurosciences: An Essential Guide. Cambridge, MA: The MIT Press.
Geschwind, D. H., Sowinski, J., Lord, C., Iversen, P., Shestack, J., Jones, P., Ducat, L., & Spence, S. J. (2001). The autism genetic resource exchange: a resource for the study of autism and related neuropsychiatric conditions. American Journal of Human Genetics, 69, 463–466.
Gingrich, J. A., & Hen, R. (2001). Dissecting the role of the serotonin system in neuropsychiatric disorders using knockout mice. Psychopharmacology (Berl), 155, 1–10.
Gordon, C. T., State, R. C., Nelson, J. E., Hamburger, S. D., & Rapoport, J. L. (1993). A double-blind comparison of clomipramine, desipramine, and placebo in the treatment of autistic disorder. Archives of General Psychiatry, 50, 441–447.
Herrero, J., Al-Shahrour, F., Diaz-Uriarte, R., Mateos, A., Vaquerizas, J. M., Santoyo, J., & Dopazo, J. (2003). GEPAS: A web-based resource for microarray gene expression data analysis. Nucleic Acids Research, 31, 3461–3467.
Kakiuchi, C., Iwamoto, K., Ishiwata, M., Bundo, M., Kasahara, T., Kusumi, I., Tsujita, T., Okazaki, Y., Nanko, S., Kunugi, H., Sasaki, T., & Kato, T. (2003). Impaired feedback regulation of XBP1 as a genetic risk factor for bipolar disorder. Nature Genetics, 35, 171–175.
Kerr, M. K., & Churchill, G. A. (2001). Bootstrapping cluster analysis: assessing the reliability of conclusions from microarray experiments. Proceedings of the National Academy of Sciences of the United States of America, 98, 8961–8965.
Liu, J., Nyholt, D. R., Magnussen, P., Parano, E., Pavone, P., Geschwind, D., Lord, C., Iversen, P., Hoh, J., Ott, J., & Gilliam, T. C. (2001). A genomewide screen for autism susceptibility loci. American Journal of Human Genetics, 69, 327–340.
Lord, C., Pickles, A., McLennan, J., Rutter, M., Bregman, J., Folstein, S., Fombonne, E., Leboyer, M., & Minshew, N. (1997). Diagnosing autism: Analyses of data from the Autism Diagnostic Interview. Journal of Autism and Developmental Disorders, 27, 501–517.
Lord, C., Risi, S., Lambrecht, L., Cook, E., Leventhal, B., DiLavore, P., Pickles, A., & Rutter, M. (2000). The autism diagnostic observation schedule-generic: a standard measure of social and communication deficits associated with the spectrum of autism. Journal of Autism and Developmental Disorders, 30, 205–223.
Lord, C., Rutter, M., & Le Couteur, A. (1994). Autism Diagnostic Interview-Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. Journal of Autism and Developmental Disorders, 24, 659–685.
Lu, A., Tang, Y., Ran, R., Clark, J. F., Aronow, B. J., & Sharp, F. R. (2003). Genomics of the periinfarction cortex after focal cerebral ischemia. Journal of Cerebral Blood Flow and Metabolism, 23, 786–810.
Luiselli, J. K., Blew, P., Keane, J., Thibadeau, S., & Holzman, T. (2000). Pharmacotherapy for severe aggression in a child with autism: “open label” evaluation of multiple medications on response frequency and intensity of behavioral intervention. Journal of Behavior Therapy and Experimental Psychiatry, 31, 219–230.
Luo, Z., & Geschwind, D. H. (2001). Microarray applications in neuroscience. Neurobiology of Disease, 8, 183–193.
Mukherjee, S., Tamayo, P., Rogers, S., Rifkin, R., Engle, A., Campbell, C., Golub, T. R., & Mesirov, J. P. (2003). Estimating dataset size requirements for classifying DNA microarray data. Journal of Computational Biology, 10, 119–142.
Philippe, A., Guilloud-Bataille, M., Martinez, M., Gillberg, C., Rastam, M., Sponheim, E., Coleman, M., Zappella, M., Aschauer, H., Penet, C., Feingold, J., Brice, A., & Leboyer, M. (2002). Analysis of ten candidate genes in autism by association and linkage. American Journal of Medical Genetics, 114, 125–128.
Pickles, A., Bolton, P., Macdonald, H., Bailey, A., Le Couteur, A., Sim, C., & Rutter M. (1995). Latent-class analysis of recurrence risks for complex phenotypes with selection and measurement error: a twin and family history study of autism. American Journal of Human Genetics, 57, 717–726.
Posey, D. I., Litwiller, M., Koburn, A., & McDougle, C. J. (1999). Paroxetine in autism. Journal of American Academy of Child and Adolescent Psychiatry, 38, 111–112.
Posey, D. J., & McDougle, C. J. (2000). The pharmacotherapy of target symptoms associated with autistic disorder and other pervasive developmental disorders. Harvard Review of Psychiatry, 8, 45–63.
Purcell, A. E., Jeon, O. H., Zimmerman, A. W., Blue, M. E., & Pevsner, J. (2001). Postmortem brain abnormalities of the glutamate neurotransmitter system in autism. Neurology, 57, 1618–1628.
Rea, M. A., Gregg, J. P., Qin, Q., Phillips, M. A., & Rice, R. H. (2003). Global alteration of gene expression in human keratinocytes by inorganic arsenic. Carcinogenesis, 24, 747–756.
Risch, N., Spiker, D., Lotspeich, L., Nouri, N., Hinds, D., Hallmeyer, J., Kalaydjieva, L., McCague, P., Dimiceli, S., & Pitts, T. (1999). A genomic screen of autism: Evidence for a multilocus etiology. American Journal of Human Genetics, 65, 493–507.
Robinson, P. D., Schutz, C. K., Macciardi, F., White, B. N., & Holden, J. J. (2001). Genetically determined low maternal serum dopamine beta-hydroxylase levels and the etiology of autism spectrum disorders. American Journal of Medical Genetics, 100, 30–36.
Schadt, E. E., Monks, S. A., Drake, T. A., Lusis, A. J., Che, N., Colinayo, V., Ruff, T. G., Milligan, S. B., Lamb, J. R., Cavet, G., Linsley, P. S., Mao, M., Stoughton, R. B., & Friend, S. H. (2003). Genetics of gene expression surveyed in maize, mouse and man. Nature, 422, 297–302.
Tang, Y., Glauser, T. A., Gilbert, D. L., Hershey, A. D., Privitera, M. D., Ficker, D. M., Szaflarski, J. P., & Sharp, F. R. (2004). Valproic acid blood genomic expression patterns in children with epilepsy – a pilot study. Acta Neurologica Scandinavica, 109, 159–168.
Tang, Y., Nee, A. C., Lu, A., Ran, R., & Sharp, F. R. (2003). Blood genomic expression profile for neuronal injury. Journal of Cerebral Blood Flow and Metabolism, 23, 310–319.
Unger, M. A., Rishi, M., Clemmer, V. B., Hartman, J. L., Keiper, E. A., Greshock, J. D., Chodosh, L. A., Liebman, M. N., & Weber, B. L. (2001). Characterization of adjacent breast tumors using oligonucleotide microarrays. Breast Cancer Research, 3, 336–341.
Warren, R. P., Margaretten, N. C., Pace, N. C., & Foster, A. (1986). Immune abnormalities in patients with autism. Journal of Autism and Developmental Disorders, 16, 189–197.
World Health Organization (1993). The ICD-10 classification for mental and behavioral disorders: diagnostic criteria for research ed. Geneva, Switzerland: World Health Organization.
Yonan, A. L., Alarcon, M., Cheng, R., Magnusson, P. K., Spence, S. J., Palmer, A. A., Grunn, A., Juo, S. H., Terwilliger, J. D., Liu, J., Cantor, R. M., Geschwind, D. H., & Gilliam, T. C. (2003). A genomewide screen of 345 families for autism-susceptibility loci. American Journal of Human Genetics, 73, 886–897.
Zien, A., Aigner, T., Zimmer, R., & Lengauer, T. (2001). Centralization: a new method for the normalization of gene expression data. Bioinformatics, 17(Suppl 1), S323–S331.
Acknowledgments
This work was supported by the MIND Institute Biomarkers Initiative, MIND Institute Genomics Core, and grant number PO1 ES 11269 from the NIEHS and US EPA. The authors would like to thank AGRE for providing cells lines and information on families used in this study. The authors express their appreciation to Zeljka Smit-McBride for culturing the AGRE cell lines, to Dawn Milliken for performing the gene expression microarray work, and to Clifford Tepper and Wenting Zhang for helpful commentary during manuscript preparation. We gratefully acknowledge the resources provided by the Autism Genetic Resource Exchange (AGRE) Consortium and the participating AGRE families. The Autism Genetic Resource Exchange is a program of Cure Autism Now and is supported, in part, by grant MH64547 from the National Institute of Mental Health to Daniel H. Geschwind (PI).
Author information
Authors and Affiliations
Corresponding author
Additional information
The authors Colin A. Baron, Stephenie Y. Liu contributed equally to this work.
Rights and permissions
About this article
Cite this article
Baron, C.A., Liu, S.Y., Hicks, C. et al. Utilization of Lymphoblastoid Cell Lines as a System for the Molecular Modeling of Autism. J Autism Dev Disord 36, 973–982 (2006). https://doi.org/10.1007/s10803-006-0134-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10803-006-0134-x