
Abstract
Oxygen evolution in harsh high acidic condition is a challenge to both zinc electrowinning and hydrogen production. In the present work, the carbon nanotubes (CNTs) sheathed in lead hybrid anode has been fabricated through electrochemical codeposition, and its electrochemical performances under zinc electrowinning condition have been discussed extensively. It is also the first time to coat CNTs by Pb with tunable method via metal oxide. The CNTs/Pb composite anode shows ten times larger exchange current density and electrochemically active surface areas (ECSA) than those of pure Pb, which indicates an extra high electro-catalytic activity. The lower overpotential and polarization resistance of the CNTs/Pb composite anode for oxygen evolution are due to integrity for efficient charge transport between matrix and PbO2 by penetrating through PbO layer. Under industrial condition of current density 500 A m−2, the overpotential can be decreased by about 120 mV compared with pure Pb during the long-term durability test. Moreover, the hybrid anode comprising ~ 0.6 wt% CNTs is also cost effective. The CNTs/Pb composite anode can be a promising and cost-effective anode in harsh high acidic condition and potentially applied in zinc electrowinning practice due to high hardness and scalable production.
Graphical abstract

Similar content being viewed by others


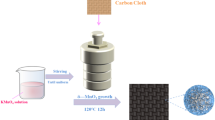
Avoid common mistakes on your manuscript.
1 Introduction
Electrowinning processing has been used commercially for metal recovery such as zinc, copper, nickel, cobalt, as well as manganese and other metals for over 100 years. In practice, lead or lead–silver is widely used as insoluble anodes owing to their high current carrying capacity, corrosion, and wear resistance in harsh working environments [1]. As lead anode has disadvantages such as elevated energy consumption [2] and consumption of Ag, thus many attempts have been made to alloy or coat the anode. Binary, ternary, or quaternary lead alloys formed by insignificant amounts of alkali metals (Li, K), the alkali earth metals (Ca, Ba, Sr), Ti, Sn, Bi, Co, Pt, or Ag can greatly improve the corrosion resistance and decrease the oxygen overpotential [3, 4]. Lead-based composite anodes containing metal oxide particles such as MnO2 [5] and IrOx [6], have exhibited better catalytic activity and preferable corrosion resistance compared with pure Pb and Pb–Ag anodes.
The oxygen evolution reaction (OER) in water oxidation is also one of the most important reactions and a bottleneck for the efficient and cost-effective production of hydrogen, since the OER is sluggish kinetically during the proton-coupled four-electron transfer steps [7, 8]. While iridium and ruthenium oxides are well-known water oxidation catalysts (WOCs) thanks to their low overpotential for OER, these noble metal-based catalysts have intrinsic drawbacks, such as source scarcity, high cost, and rapid deterioration [9, 10]. Earth-abundant OER electrocatalysts based on first-row 3d transition metals were newly developed and led to substantial advancements, which has comparable or superior catalytic activities to the noble metal-based materials [11,12,13,14]. However, most of these transition metals are effective under basic or nearly neutral pH conditions. A few catalysts [9, 15,16,17,18,19,20,21] are efficacious for the OER at low pH with potential applications both in artificial photosynthesis systems and in more conventional polymer electrolyte membrane electrolysers [22]. These promising OER electrocatalysts in acidic electrolyte usually work well at a small current density (~ 10 mA cm−2), and in < 1 mol L−1 H2SO4, which is inadequate for the industrial zinc electrowinning process.
Recently, as the low-cost metal-free catalysts, doped carbon nanomaterials, such as surface-oxidized and electrochemically activated multiwall carbon nanotubes (MWCNTs) [23], laser-induced graphene [24], and nitrogen-doped graphene mesh [25], have been concerned for OER [26]. In these materials, oxygen containing functional groups and high catalytic nanoparticles play significant roles as the catalytic active sites for OER. Nevertheless, carbon will be oxidized to CO2 during the OER process, which will lead to the function collapse as either the catalyst support or the active catalyst [27,28,29].
In this contribution, inorganic/CNTs composites were strongly coupled as effective electrocatalysts for OER in the zinc electrowinning process. This composite shows excellent performances in both catalytic activity and durability. To the best of our knowledge, it is the first report of CNTs sheathed in metal as electrocatalysts. Being different from the previous publications of CNTs decorated by nanoparticles [30, 31], CNTs in the present work was uniformly coated with metal, which can decrease the overpotential by maintaining structural integrity for efficient charge transport. Moreover, the lead coating can separate CNTs from harsh corrosion environment for prolonging the practical service life. This method with in situ nucleation by co-electrodeposition was also scalable and cost effective for potential applications, and can also provide a guideline to cover CNTs with metals for other purposes.
2 Experimental
2.1 Chemicals and materials
Zinc sulfide (ZnSO4·7H2O), lead acetate trihydrate (Pb(CH3COO)2·3H2O), ammonium acetate (CH3COONH4), boric acid (H3BO3), glycerol, sodium dodecylbenzenesulfonate (SDBS), polyvinylpyrrolidone (PVP, K30), sulfuric acid (H2SO4) were of analytical grade and purchased from Sinopharm Chemical Reagent Co. Ltd. The chemicals were used as received without further purification. Lead foil (99+ %) and Ti foil (99.7+ %) were purchased from Alfa Aesar. MWCNTs with average diameter of 7–11 nm and length of 5–20 µm were provided by Cnano Technology Ltd. High-purity water (18 M Ω) was used throughout the experiments.
2.2 Synthesis of CNTs/Pb powders
CNTs/Pb powders were fabricated by electrochemical codeposition under ultrasonic assistance (25/40 kHz, 120 W). The electrolyte was composed of 2 mol L−1 CH3COONH4, 0.005 mol L−1 lead acetate, 0.1 mol L−1 H3BO3, 0.6 mol L−1 glycerol, 0.5 g L−1 SDBS, 0.5 g L−1 PVP, and 0.5 g L−1 CNTs. At first, the SDBS, PVP, and CNTs were mixed in water together under ultrasonication for 30 min to produce a homogeneous suspension. Then, other reagents were added into the suspension to obtain the electrolyte with a pH value of ~ 6.8. An UltraBasic pH Meter (UB-7, Denver) was used to determine the pH values.
Electrochemical codeposition of CNTs/Pb was carried out at galvanostatic mode with a cathodic current density 5 mA cm−2 at room temperature under ultrasonic condition. A titanium plate and a pure lead foil were employed as the cathode and anode, respectively. During the electrodeposition, CNTs/Pb powders were scraped from the cathode, and thoroughly rinsed with pure water and ethanol, and then dried in a vacuum oven at 60 °C overnight. Pure lead powders were prepared with the same process as above without the introduction of CNTs.
2.3 Preparation of CNTs/Pb anode
The as-deposited CNTs/Pb powders were consolidated by cold pressing with an applied pressure of 413 MPa for 10 min. Then, the composite was annealed at 250 °C under vacuum with a pressure < 0.1 kPa for 2 h. Subsequently, cold pressing was applied again with a pressure of 413 MPa for 10 min. At the same time, the pure lead powder anode was fabricated with the same process above for the control experiment. All anodes for electrochemical measurement were polished with wet abrasive papers grade by grade to 2000 grit, and then ultrasonically cleaned with ethanol and acetone. The polished samples were tested immediately.
2.4 Characterization of materials
The content of CNTs in CNTs/Pb anode was determined by C/S analyzer (Eltra, CS 2000). X-ray diffraction (XRD, Rigaku D/Max 2200) was carried out using Cu Kα (1.54056 Å) target in steps of 0.02° with a scan speed of 8° min−1. The morphological structures of the powders were investigated using scanning electron microscopy (FEI, Quanta 200) and field-emission transmission electron microscopy (FEI, Tecanai G2 TF30). Both the atomic compositions and structure were determined by energy-dispersive spectrometry (EDS) with scanning transmission electronic microscope (STEM) and high-resolution transmission electronic microscope (HRTEM). Raman spectroscopy (LabRAM, HR Evolution) was used to identify the existence of CNTs with He–Cd Laser (λ = 532 nm). HV hardness values of anodes were examined using a micro Vickers hardness tester (HXD-1000TMC/LCD), with a test force of 10 gf and duration time of 10 s.
2.5 Electrochemical measurements
The electrochemical characterizations were performed on a computer-controlled Versastat 3 potentiostat/galvanostat (Ametek, USA) by VersaStudio software with a three-electrode system in simulating electrolyte solution of zinc electrowinning which consisted of 60 g L−1 (= 0.917 M) Zn2+ and 160 g L−1 (= 1.633 M) H2SO4. The temperature of the electrolyte was controlled at 25 °C by a thermostatic water bath (Yuhua, DF101). The cell was kept in the dark to avoid the influence of photo-electrochemical reaction of the electrode. Three different anodic materials were used as the working electrodes, which were sealed tightly in electrode hold (Ida, JJ210) with an exposure area of 1 cm2. A platinum gauze and mercury/mercurous sulfate (in sat’d K2SO4) (MSE, 0.652V vs. RHE) were used as the counter electrode and the reference electrode, respectively. The potential values in the present work were converted to reversible hydrogen electrode (RHE) scale through the following Eq. (1) unless otherwise stated [20, 23]:
All overpotentials (η) for the oxygen evolution reaction (OER) from H2O were calculated by the Eq. (2) [20]:
where i is the current density, Rs is the solution resistance determined by electrochemical impedance spectroscopy (EIS), \(E_{{{{\text{H}}_2}{\text{O}}/{{\text{O}}_2}}}^{'}\) is the thermodynamic oxygen evolution potential in acid and defined as 1.229 − 0.059 × pH (V).
The pretreatment of anodes using consecutive multiple cyclic voltammetry (CV) was performed 30 runs from 1 to 2.2 V with a sweep rate of 30 mV s−1 to obtain consistent and reproducible results [32]. The CV measurements were conducted from 1 to 2.4 V with a sweep rate of 10 mV s−1. The linear sweeping voltammograms (LSVs) were recorded by a linear sweep of potential at a sweep rate of 1 mV s−1 from 1 to 2.4 V in the region of both oxidation of anode and oxygen evolution. The slow sweep rate was chosen to minimize any pseudocapacitance and reach steady-state current density. The solution was rigorously stirred with a Teflon stir bar to keep the reaction in the activation-controlled region and avoid mitigating mass transport effects [20]. Tafel plots were obtained by plotting overpotential (η) versus Log (i) from LSVs, where i is current densities based on the geometrical surface areas of the electrodes. Electrochemical impedance spectroscopy (EIS) measurements were conducted at a potential of 2.152 V where oxygen bubbles begin to evolve intensely. The amplitude of the sinusoidal wave was 10 mV, and the frequency scan range was from 100 kHz to 0.1 Hz. Impedance data were fitted to the equivalent circuit using the Zsimpwin software (EG&G). The best fit was obtained by minimizing the χ2 (chi squared).
3 Results and discussion
To prepare powders of CNTs sheathed in Pb, the pH of electrolyte must be controlled to favor forming PbO based on the potential–pH equilibrium diagram of lead–water system [33]. Figure 1a–c shows synthesis mechanism of the CNTs/Pb powder, and Fig. 1d is oxygen evolution on the surface of CNTs/Pb composite anode with a sandwich structure. Firstly, Pb2+ will be adsorbed on the surface of CNTs to form in situ hydrous amorphous PbO layer as homogeneous nucleation process (as in Fig. 1a). When they contact the cathode, PbO inside will begin to be reduced and O atoms will transfer outside due to electrons exchange (as in Fig. 1b, c).

Schematic illustration of preparation of CNTs-coated Pb as anode for water oxidation. a In situ PbO nucleation on CNTs; b, c electrochemical reduction of PbO to Pb; d oxygen evolution with short pass PbO layer with CNTs
3.1 Physical characterization
The as-deposited powders were investigated by XRD and the results were shown in Fig. 2. The diffraction patterns show that both Pb metal and PbO co-existed in the powder products. Both of them are with the highly preferred orientation on (111), which is the most stable plane in face-centered cubic metals [34].

XRD patterns of CNTs sheathed in lead powders
Raman spectroscopy is a convenient and useful method for characterization of carbon nanotube. As shown in Fig. 3a, D-band located around 1342 cm−1 and a G-band around 1588 cm−1 were observed in the as-synthesized CNTs/Pb powder. The higher order peak appeared at 2692 cm−1 and a small broad peak at 2933 cm−1 can be assigned to a combination of D + D and D + G bands, respectively, as is shown in Fig. 3a. The G-band is a characteristic for the bond stretching of sp2 bonded pairs, while the D-band is assigned to the sp3 defect sites [35]. ID/IG can be used to characterize the degree of organization of CNTs. The ratios of ID/IG for CNTs/Pb and pristine CNTs are 0.95 and 1.14, respectively, which means that the defect sites of pristine CNTs have been covered by Pb metal. In other words, Pb is preferentially deposited in the defect sites of CNTs, which consist with the fact that both the D-band and G-band peaks of CNTs/Pb have shown an obvious blue shift, as is displayed in Fig. 3b. The peak in 142 cm−1 can be contributed to PbO [36], which agrees well with the results of XRD.

Raman spectra of CNTs sheathed in lead powders and pristine CNTs (a) and Raman spectra in the range of 1000–1800 cm−1 showing ID/IG (b)
The as-deposited powders were investigated by both SEM and TEM, and the results are shown in Fig. 4. As is shown in Fig. 4a, the CNTs-coated Pb shows well-defined edges and similar diameters of the pristine ones, and are uniformly distributed without large agglomeration. In addition, it shows no obvious destruction or shearing effect on CNTs caused by ultrasonic. The in situ nucleation process of Pb on the surface of CNTs is preferable.

SEM of CNTs sheathed in lead (a); HRTEM image (b) and selected area diffraction pattern (c) of CNTs/Pb powders; structural models shown in the right side of (c), and high-angle annular dark-field scanning transmission electron microscopy-energy-dispersive (d)
To further identify the composition and structure of Pb on the surface of CNTs, HRTEM was carried out to observe the interface between CNTs and Pb, and the results are shown in Fig. 4b. The EDS analysis (Fig. 4d) shows that only elements of lead and carbon exist in the powder product and copper belongs to the copper mesh applied during the EDS analysis. Oxygen is out of determination owing to its small quantity. The coating presents a core–shell structure with Pb existing inside and PbO as the shell. Both of them have the similar crystal structures, and there is no obvious boundary between the phases, owing to the fact that the O atoms in PbO migrate outwards as a result of the electric field force in the cathode. There is also no clear boundary in the interfaces between the CNT and Pb or PbO, indicating the existence of bond-joint between them (as the red circles A and B in Fig. 4b). Figure 4c shows the HRTEM patterns of the CNTS/Pb powder. The interspacing distances of 0.310 and 0.285 nm correspond to the (111) planes of PbO and Pb (as in the right insets of Fig. 4c), respectively, indicating the preferred orientation on (111) plane of these two phases. This conclusion also agrees well with the results of both XRD and Raman spectra.
High-angle annular dark-field scanning transmission electron microscopy–energy-dispersive X-ray spectroscopy (HAADF–STEM–EDS) line scan analysis is shown in Fig. 5. A single CNT-coated Pb in SEM shows a uniform distribution of Pb layer throughout the whole CNT in Fig. 5a. EDS in Fig. 5b confirms the elements of C and Pb, and element distribution across the CNTs in Fig. 5c indicates that the thickness of the Pb coating is about 5 nm.

High-angle annular dark-field scanning transmission electron microscopy–energy-dispersive X-ray spectroscopy (HAADF–STEM–EDS) line scan analysis of a single CNT-coated Pb. a SEM of the CNT; b EDS of the CNT; c Line scan analysis across the CNT with element distribution of C, Cu, and Pb in weight (along the red line in a)
3.2 Electrochemical performance
Electrocatalysis of CNTs/Pb composition anode (abbreviated as “CNTs/Pb” hereafter) was investigated and compared with anodes made of pure Pb powders and pure Pb foil (abbreviated, respectively, as “Powder Pb” and “Pure Pb” hereafter).
CVs recorded for Pure Pb, Powder Pb, and CNTs/Pb electrodes in a potential range between 1 and 2.4 V at the sweep rate of 10 mV s−1 are shown in Fig. 6a. The CVs are in excellent agreement with our previous report [37] and other literatures [4, 38]. Peak a at 1.65 V is attributed to the oxidation of Pb, while peak b at 1.93 V is due to the oxidation of PbSO4 in the outer layer to PbO2 in sulfuric acid solution. Cathodic CV peak c observed at about 1.51 V during the reverse scan is assigned to the reduction of PbO2 to PbSO4. The reduction charge (Q) for the three cycles was calculated from the CVs to evaluate the catalytic activity of the lead anode [39]. The ratio of Q(CNTs/Pb)/Q(Pure Pb) is 1.38 and almost the same as that of Q(Powder Pb)/Q(Pure Pb). Therefore, it can be concluded that both CNTs/Pb and Powder Pb are much more active than that of Pure Pb.

a Cyclic voltammograms recorded at 10 mV s−1; b linear sweeping voltammograms at 1 mV s−1 and Tafel plots of oxygen evolution (inset); c Nyquist plots and the equivalent circuit used for data analysis for the impedance data recorded (inset); d chronopotentiometric curves of current density of 500 A m−2 for the CNTs/Pb (triangle), Powder Pb (circle), and Pure Pb (diamond) in the zinc electrowinning electrolyte at 25 °C
LSV and Tafel plots can be used to further investigate the activity of the anodes (Fig. 6b). At a current density of 10 mA cm−2, the potential for oxygen evolution is 2.094 V for the CNTs/Pb, whereas those are 2.145 V, and 2.198 V for Powder Pb and Pure Pb, respectively. The overpotential of OER on CNTs/Pb decreased by ~ 100 mV compared to Pure Pb. The inset of Fig. 6b is Tafel plots of OER which usually presented two distinct linear segments in both the low-potential region and high-potential region due to partially evolved O3 [40, 41]. The Tafel formula was calculated for the exchange current density (i0) in the low-potential region, and the results are shown in Table 1. The Tafel slops (b1) of CNTs/Pb, Powder Pb, and Pure Pb all are ~ 170 mV decade−1, and the β-values (anodic transfer coefficient) are similar, which indicated the rate determining steps of OER for all anodes are almost the same. Tafel slops of OER were usually high in acidic condition and with large overpotential [42]. Slopes greater than 120 mV decade−1 are also attributed to the turnover-limiting chemical step and quasi-equilibrium [20]. While the overpotential increases continually, OER of all anodes will be nearly equal and independent of electrode. The exchange current density (i0) of CNTs/Pb is as much as ten times larger than that of pure Pb, which suggests an excellent electrocatalysis for OER owing to a large electrochemically active surface area [43].
When the potential increases to 2.152 V which is much higher than onset potential of CNTs/Pb, Powder Pb, and Pure Pb, the current densities of the three anodes are 19 mA cm−2, 12 mA cm− 2, and 5 mA cm−2, respectively. This potential is a nice window for EIS due to both oxygen bubbling intensely and impedance requirements. The Nyquist plots of three anodes at the potential (Fig. 6c) show electrochemically controlled reaction with a highly active surface electrode thanks to typical RC characters and small circles. The well-known Randle’s circuit (inset in Fig. 6c) is used as the equivalent circuit to fit the data. Here, Rs is the solution (electrolyte) resistance, and Rp is the polarization (or charge transfer) resistance for oxygen evolution. In addition, the constant phase element (CPE, Qdl) is used for double-layer capacitance at the Pb|PbO2|electrolyte interface due to the fractal morphology. The CPE admittance, Y, has an expression,
where n is the frequency dependence of the CPE admittance (Y) with its value between 0 and 1, ω is the angular velocity defined as 2πf with f being frequency, and YQ is a frequency independent of the CPE admittance. When n = 1, the CPE is a pure capacitance. The Cdl values can be calculated from the constant phase element, Qdl, using a relation [43, 45]:
The parameters of the equivalent circuit for CNTs/Pb, Powder Pb, and Pure Pb were calculated and listed in Table 2. The Rs values of all anodes are about as high as 2.3 Ω cm2 since the oxygen bubbles on the surface blocked the interface of electrode/electrolyte, owing to the large current density. The value of n is smallest for CNTs/Pb as a result of large dispersion. It is well known that n has a relation with the fractal geometry (D: between 2 for an absolutely flat dimension and 3 dimension) as n = 1/(D-1) [46]. Thus, it is a reasonable n value for a composite material (CNTs/Pb) compared with pure metals (Pb). The double-layer capacitance (C) of CNTs/Pb (0.017 F cm−2) is one order of magnitude lager than that of Pure Pb (0.0016 F cm−2), which shows an extra high electrochemically active surface areas (ECSA) [47] and agrees excellently with the exchange current densities (i0) of Tafel’s results. The Rp value of CNTs/Pb is close to one-third that of Pure Pb which indicates the higher active catalysis for OER. The Pb cations are octahedrally surrounded by the oxygen anions in lattice. The conduction band is mainly due to the interaction of the t2g orbital of t2g cation and the p-orbital of the oxygen anion. The sp2 state of CNTs is not distorted throughout the structure during the plating procedure. The π-electrons are delocalized and transferred between unoccupied p-orbital of PbO2 and Pb metal.
PbO2 is a degenerated n-type semiconductor possessing a concentration of defects as high as ~ 1022 cm−3 [48], which makes PbO2 quite similar to metals (conductivity of 59 S m−1). However, the PbO layer between PbO2 and Pb is a typical n-type semiconductor with a forbidden energy gap of 2.66 eV and high ohmic resistivity (~ 1012 Ω cm) and blocks the transfer of electrons. CNTs is metallic with a high conductivity of ~ 100 S m−1, and thus can bridge between the PbO2 and Pb to shorten PbO layer (as in Fig. 1d), and result in a low Rp value. Xu et al. also reported that CNTs could support a long-range electron exchange between the Pt and Pd nanoparticles without direct atomic contact [49].
The catalytic stability of CNTs/Pb, Powder Pb, and Pure Pb was assessed with chronopotentiometric curves at 500 A m−2 for 10 h, which was a typical current density value in zinc electrowinning, as shown in Fig. 6d. The overpotential of CNTs/Pb anode is very stable during test and decreased by about 120 mV and 70 mV, respectively, compared with Pure Pb and Powder Pb. It is well known that it will save about 95 kW h electrical powder per ton zinc when overpotential decreases by 100 mV.
The morphological structures of CNTs/Pb anode before and after the stability measurement are shown in Fig. 7. It is shown that CNTs-coated Pb particles stand out of the matrix, owing to grinding of the anode (as in Fig. 7a, b). After 10-h electrolysis, β-PbO2 particles with diameters of about 2 µm uniformly covered on the surface, which is the same as Pb-based anodes. The content of CNTs in the CNTs/Pb composite is about 0.60 wt%, which suggests the anode is cost effective. The Vickers hardness of CNTs/Pb is tested to be 49.1 Hv, which is about four times larger than that of Pure Pb. The electrochemical performances, along with the deformation reduction properties, suggest the potential application of the CNTs/Pb composites in industry.

Morphology of CNTs/Pb anode before (a, b) and after (c) durability measurement for 10 h in the zinc electrowinning electrolyte at 25 °C
4 Conclusion
In summary, CNTs sheathed in Pb was prepared by electrochemical codeposition method based on the Pourbaix diagram. CNTs are uniformly coated Pb via metal oxide with the thickness of ~ 5 nm. The CNTs/Pb composite anode shows ten times larger exchange current density and electrochemically active surface areas (ECSA) than Pure Pb, which indicates an extra high electro-catalytic activity. The low overpotential and polarization resistance of the CNTs/Pb composite for oxygen evolution are due to bridging the interfacial interactions which shorten the electron pathway. The CNTs/Pb composite with long-term durability in harsh high acidic condition can be a promising and cost-effective anode in zinc electrowinning thanks to high hardness and scalable production.
References
Free ML, Moats M, Houlachi G, Asselin E, Allanore A, Yurko J, Wang S (2012) Electrometallurgy, 1 edn. Wiley, New York
Loutfy RO, Leroy RL (1978) Energy efficiency in metal electrowinning. J Appl Electrochem 8(6):549–555. https://doi.org/10.1007/bf00610801
Petrova M, Noncheva Z, Dobrev T, Rashkov S, Kounchev N, Petrov D, Vlaev S, Mihnev V, Zarev S, Georgieva L, Buttinelli D (1996) Investigation of the processes of obtaining plastic treatment and electrochemical behaviour of lead alloys in their capacity as anodes during the electroextraction of zinc. Hydrometallurgy 40(3):293–318. https://doi.org/10.1016/0304-386X(95)00010-E
Hrussanova A, Mirkova L, Dobrev T (2004) Influence of additives on the corrosion rate and oxygen overpotential of Pb-Co3O4, Pb-Ca-Sn and Pb-Sb anodes for copper electrowinning. Hydrometallurgy 72(3–4):215–224. https://doi.org/10.1016/j.hydromet.2003.07.005
Ma R, Cheng S, Zhang X, Li S, Liu Z, Li X (2016) Oxygen evolution and corrosion behavior of low-MnO2-content Pb-MnO2 composite anodes for metal electrowinning. Hydrometallurgy 159:6–11. https://doi.org/10.1016/j.hydromet.2015.10.031
Le Pape-Rerolle, C, Petit MA, Wiart R (1999) Catalysis of oxygen evolution on IrOx/Pb anodes in acidic sulfate electrolytes for zinc electrowinning. J Appl Electrochem 29(11):1347–1350
Grimaud A, Diaz-Morales O, Han B, Hong WT, Lee Y-L, Giordano L, Stoerzinger KA, Koper MTM, Shao-Horn Y (2017) Activating lattice oxygen redox reactions in metal oxides to catalyse oxygen evolution. Nat Chem 9(5):457–465. https://doi.org/10.1038/nchem.2695
Chen S, Thind SS, Chen A (2016) Nanostructured materials for water splitting: state of the art and future needs. Electrochem Commun 63:10–17. https://doi.org/10.1016/j.elecom.2015.12.003
Sardar K, Petrucco E, Hiley CI, Sharman JDB, Wells PP, Russell AE, Kashtiban RJ, Sloan J, Walton RI (2014) Water-splitting electrocatalysis in acid conditions using ruthenate-iridate pyrochlores. Angew Chem Int Ed 53(41):10960–10964. https://doi.org/10.1002/anie.201406668
Cherevko S, Geiger S, Kasian O, Mingers A, Mayrhofer KJJ (2016) Oxygen evolution activity and stability of iridium in acidic media. J Electroanal Chem 773:69–78. https://doi.org/10.1016/j.jelechem.2016.04.033
Zheng Y, Jiao Y, Zhu Y, Cai Q, Vasileff A, Li LH, Han Y, Chen Y, Qiao S-Z (2017) Molecule-level g-C3N4 coordinated transition metals as a new class of electrocatalysts for oxygen electrode reactions. J Am Chem Soc 139(9):3336–3339. https://doi.org/10.1021/jacs.6b13100
Wan S, Qi J, Zhang W, Wang W, Zhang S, Liu K, Zheng H, Sun J, Wang S, Cao R (2017) Hierarchical Co(OH)F superstructure built by low-dimensional substructures for electrocatalytic water oxidation. Adv Mater 29(28):1700286. https://doi.org/10.1002/adma.201700286
Tung C-W, Hsu Y-Y, Shen Y-P, Zheng Y, Chan T-S, Sheu H-S, Cheng Y-C, Chen HM (2015) Reversible adapting layer produces robust single-crystal electrocatalyst for oxygen evolution. Nat Commun 6:8106. https://doi.org/10.1038/ncomms9106
Wang H, Lee H-W, Deng Y, Lu Z, Hsu P-C, Liu Y, Lin D, Cui Y (2015) Bifunctional non-noble metal oxide nanoparticle electrocatalysts through lithium-induced conversion for overall water splitting. Nat Commun 6:7261. https://doi.org/10.1038/ncomms8261
Chang SH, Danilovic N, Chang K-C, Subbaraman R, Paulikas AP, Fong DD, Highland MJ, Baldo PM, Stamenkovic VR, Freeland JW, Eastman JA, Markovic NM (2014) Functional links between stability and reactivity of strontium ruthenate single crystals during oxygen evolution. Nat Commun 5:4191. https://doi.org/10.1038/ncomms5191
Diaz-Morales O, Raaijman S, Kortlever R, Kooyman PJ, Wezendonk T, Gascon J, Fu WT, Koper MTM (2016) Iridium-based double perovskites for efficient water oxidation in acid media. Nat Commun 7:12363. https://doi.org/10.1038/ncomms12363
Seitz LC, Dickens CF, Nishio K, Hikita Y, Montoya J, Doyle A, Kirk C, Vojvodic A, Hwang HY, Norskov JK, Jaramillo TF (2016) A highly active and stable IrOx/SrIrO3 catalyst for the oxygen evolution reaction. Science 353(6303):1011–1014. https://doi.org/10.1126/science.aaf5050
Kim J, Shih P-C, Tsao K-C, Pan Y-T, Yin X, Sun C-J, Yang H (2017) High-performance pyrochlore-type yttrium ruthenate electrocatalyst for oxygen evolution reaction in acidic media. J Am Chem Soc 139(4):12076–12083. https://doi.org/10.1021/jacs.7b06808
Bloor LG, Molina PI, Symes MD, Cronin L (2014) Low pH electrolytic water splitting using earth-abundant metastable catalysts that self-assemble in situ. J Am Chem Soc 136(8):3304–3311. https://doi.org/10.1021/ja5003197
Huynh M, Bediako DK, Nocera DG (2014) A functionally stable manganese oxide oxygen evolution catalyst in acid. J Am Chem Soc 136(16):6002–6010. https://doi.org/10.1021/ja413147e
Mondschein JS, Callejas JF, Read CG, Chen JYC, Holder CF, Badding CK, Schaak RE (2016) Crystalline cobalt oxide films for sustained electrocatalytic oxygen evolution under strongly acidic conditions. Chem Mater 29(3):950–957. https://doi.org/10.1021/acs.chemmater.6b02879
Roger I, Shipman MA, Symes MD (2017) Earth-abundant catalysts for electrochemical and photoelectrochemical water splitting. Nat Rev Chem 1:0003. https://doi.org/10.1038/s41570-016-0003
Lu X, Yim W-L, Suryanto BHR, Zhao C (2015) Electrocatalytic oxygen evolution at surface-oxidized multiwall carbon nanotubes. J Am Chem Soc 137(8):2901–2907. https://doi.org/10.1021/ja509879r
Zhang J, Zhang C, Sha J, Fei H, Li Y, Tour JM (2017) Efficient water-splitting electrodes based on laser-induced graphene. ACS Appl Mater Interfaces 9(32):26840–26847. https://doi.org/10.1021/acsami.7b06727
Tang C, Wang H-F, Chen X, Li B-Q, Hou T-Z, Zhang B, Zhang Q, Titirici M-M, Wei F (2016) Topological Defects in metal-free nanocarbon for oxygen electrocatalysis. Adv Mater 28(32):6845–6851. https://doi.org/10.1002/adma.201601406
Li X, Fang Y, Zhao S, Wu J, Li F, Tian M, Long X, Jin J, Ma J (2016) Nitrogen-doped mesoporous carbon nanosheet/carbon nanotube hybrids as metal-free bi-functional electrocatalysts for water oxidation and oxygen reduction. J Mater Chem A 4(34):13133–13141. https://doi.org/10.1039/C6TA04187F
Wang W, Luo J, Chen S (2017) Carbon oxidation reactions could misguide the evaluation of carbon black-based oxygen-evolution electrocatalysts. Chem Commun 53:11556–11559. https://doi.org/10.1039/C7CC04611A
Pérez-Rodríguez S, Sebastián D, Lázaro MJ, Pastor E (2017) Stability and catalytic properties of nanostructured carbons in electrochemical environments. J Catal 355:156–166. https://doi.org/10.1016/j.jcat.2017.09.019
Ashton SJ, Arenz M (2012) Comparative DEMS study on the electrochemical oxidation of carbon blacks. J Power Sources 217:392–399. https://doi.org/10.1016/j.jpowsour.2012.06.015
Zhang Y-X, Guo X, Zhai X, Yan Y-M, Sun K-N (2015) Diethylenetriamine (DETA)-assisted anchoring of Co3O4 nanorods on carbon nanotubes as efficient electrocatalysts for the oxygen evolution reaction. J Mater Chem A 3(4):1761–1768. https://doi.org/10.1039/C4TA04641B
Lu X, Zhao C (2013) Highly efficient and robust oxygen evolution catalysts achieved by anchoring nanocrystalline cobalt oxides onto mildly oxidized multiwalled carbon nanotubes. J Mater Chem A 1(39):12053–12059. https://doi.org/10.1039/C3TA12912H
Yu P, O’Keefe TJ (1999) Evaluation of lead anode reactions in acid sulfate electrolytes. J Electrochem Soc 146(4):1361–1369. https://doi.org/10.1149/1.1391771
Maksymiuk K, Stroka J, Galus Z (2009) Chemistry, electrochemistry, and electrochemical applications. Elsevier, Amsterdam, pp 762–771. https://doi.org/10.1016/B978-044452745-5.00898-4
Watanabe T (2004) Film formation mechanism in electrodeposition. In: Watanabe T (ed) Nano plating. Elsevier, Oxford, pp 95–139. https://doi.org/10.1016/B978-008044375-1/50011-7
Sadezky A, Muckenhuber H, Grothe H, Niessner R, Pöschl U (2005) Raman microspectroscopy of soot and related carbonaceous materials: spectral analysis and structural information. Carbon 43(8):1731–1742. https://doi.org/10.1016/j.carbon.2005.02.018
Reid ES, Cooney RP, Hendra PJ, Fleischmann M (1977) A raman spectroscopic study of corrosion of lead electrodes in aqueous chloride media. J Electroanal Chem Interfacial Electrochem 80(2):405–408. https://doi.org/10.1016/S0022-0728(77)80063-6
Yang CJ, Park S-M (2013) Electrochemical behavior of PbO2 nanowires array anodes in a zinc electrowinning solution. Electrochim Acta 108(1):86–94. https://doi.org/10.1016/j.electacta.2013.06.068
Inguanta R, Piazza S, Sunseri C (2008) Growth and characterization of ordered PbO2 nanowire arrays. J Electrochem Soc 155(12):K205–K210. https://doi.org/10.1149/1.2988728
Jaimes R, Miranda-Hernández M, Lartundo-Rojas L, González I (2015) Characterization of anodic deposits formed on Pb–Ag electrodes during electrolysis in mimic zinc electrowinning solutions with different concentrations of Mn(II). Hydrometallurgy 156:53–62. https://doi.org/10.1016/j.hydromet.2015.05.008
Da Silva LM, De Faria LA, Boodts JFC (2003) Electrochemical ozone production: influence of the supporting electrolyte on kinetics and current efficiency. Electrochim Acta 48(6):699–709. https://doi.org/10.1016/S0013-4686(02)00739-9
Zhang C, Duan N, Jiang L, Xu F, Luo J (2018) Influence of Mn2+ ions on the corrosion mechanism of lead-based anodes and the generation of heavy metal anode slime in zinc sulfate electrolyte. Environ Sci Pollut Res 25(12):11958–11969. https://doi.org/10.1007/s11356-018-1443-2
Gerken JB, McAlpin JG, Chen JYC, Rigsby ML, Casey WH, Britt RD, Stahl SS (2011) Electrochemical water oxidation with cobalt-based electrocatalysts from pH 0–14: the thermodynamic basis for catalyst structure, stability, and activity. J Am Chem Soc 133(36):14431–14442. https://doi.org/10.1021/ja205647m
McCrory CCL, Jung S, Peters JC, Jaramillo TF (2013) Benchmarking heterogeneous electrocatalysts for the oxygen evolution reaction. J Am Chem Soc 135(45):16977–16987. https://doi.org/10.1021/ja407115p
Marshall AT, Vaisson-Béthune L (2015) Avoid the quasi-equilibrium assumption when evaluating the electrocatalytic oxygen evolution reaction mechanism by Tafel slope analysis. Electrochem Commun 61:23–26. https://doi.org/10.1016/j.elecom.2015.09.019
Hsu CH, Mansfeld F (2001) Concerning the conversion of the constant phase element parameter Y0 into a capacitance. Corrosion 57(9):747–748. https://doi.org/10.5006/1.3280607
Mulder WH, Sluyters JH, Pajkossy T, Nyikos L (1990) Tafel current at fractal electrodes: connection with admittance spectra. J Electroanal Chem Interfacial Electrochem 285(1–2):103–115. https://doi.org/10.1016/0022-0728(90)87113-x
McCrory CCL, Jung S, Ferrer IM, Chatman SM, Peters JC, Jaramillo TF (2015) Benchmarking hydrogen evolving reaction and oxygen evolving reaction electrocatalysts for solar water splitting devices. J Am Chem Soc 137(13):4347–4357. https://doi.org/10.1021/ja510442p
Mindt W (1969) Electrical properties of electrodeposited PbO2 films. J Electrochem Soc 116(8):1076–1080. https://doi.org/10.1149/1.2412217
Xu F, Lu Q, Ye L, Tsang E (2017) Intermix of metal nanoparticles-single wall carbon nanotubes. Chem Commun 53:7653–7656. https://doi.org/10.1039/C7CC03696E
Acknowledgements
This work was financial supported by the National Natural Science Foundation of China (Project No. 51664040 and 51304094), the Analysis and Testing Foundation of Kunming University of Science and Technology (Project No. 2016T20080042). Many thanks to Prof. Hongtao Guan and Zhengwei Xiao for suggestion of grammar.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Yang, Cj., Shen, Qf., Zhai, Dc. et al. Carbon nanotubes sheathed in lead for the oxygen evolution in zinc electrowinning. J Appl Electrochem 49, 67–77 (2019). https://doi.org/10.1007/s10800-018-1277-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10800-018-1277-0