
Abstract
Previously optimized anatase and nitrogen-doped anatase TiO2 coatings have been grown by low-frequency plasma-enhanced chemical vapor deposition (PECVD) on different kinds of substrates at low plasma power (64 W) and high plasma power (100 W) for photo-electrochemical studies. Nitrogen-doped TiO2 layers exhibit better photoactivity and also higher electronic conductivity under UV and visible irradiations than non-doped materials. The main reason is that nitrogen introduction induces TiO2 band gap tailoring towards higher wavelengths. In addition, films prepared at low plasma power present a ‘typical photo-material’ behavior (whose activity depends directly on the presence of light) while layers synthesized at higher plasma power contain an initial conductive phase giving them an activity that exists in the dark yet and can be slightly enhanced by illumination. Such conclusions are prominent in the field of photo-anodic thin films; indeed PECVD could constitute a promising approach for tailoring the efficiency of photo-electrochemical cells for hydrogen production under solar light.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The ‘Solar-to-Hydrogen’ approach has been widely discussed over the past twenty years, especially with the increase of the negative side effects derived from the excess usage of fossil fuels [1,2,3,4]. In fact, the main reasons behind hydrogen investigation as an alternative green fuel are its availability and its exceptional energetically efficient combustion generating only water as a by-product [5]. The problem remains in the fact that such a compound could not be found naturally as H2 but is chemically bounded to other elements like carbon or oxygen. For that, energy supply is necessary in order to split H2 combining products, thus producing hydrogen as a fuel from a primary source.
Many works have been performed in order to find convenient hydrogen primary sources with good gas yields. As a first trial, thermochemical processes have been presented for hydrogen production from fossil fuels. J. Jechura presented the process of H2 generation from natural gas by steam methane reforming (SMR) [6]. A range of 95–98% hydrogen purity was obtained but, even though the process is cheap, the SMR-integrated systems still require high maintenance. Coal gasification has also been reported in various studies [7,8,9,10]. Still such techniques require high-energy initial supplies, they generate carbonated compounds particularly carbon dioxide (CO2) as by-products and the produced H2 is obtained with low purity percentages. Biological processes, taking advantages of the microorganisms’ capacities in biomass digestion for H2 release, have also been investigated in the field of hydrogen generation [11,12,13]. Despite the fact that these investigations are very new and present good gas yield perspectives, the processes preparation and manipulation conditions remain delicate and sensitive [14]. Further studies should be done in order to improve many aspects of such processes, including low-cost materials and high-efficiency microbes.
The previously cited investigations have been based on compounds that initially do not include hydrogen but whose reaction generates this energetic gas as a by-product. Then, the concept of ‘Hydrogen generation by water splitting’ emerged from the fact that H2O, the most available compound on earth, could be dissociated into its elements, oxygen and hydrogen. For that, water electrolysis using external electrical supply has been studied and optimized under several experimental conditions in order to conclude on the best materials performances [14,15,16,17,18].
In a water-splitting system, oxidation occurs on the anode surface (linked to the positive pole of the external power source) generating oxygen, and reduction occurs on the cathode surface (linked to the negative pole of the external source) generating hydrogen. The most well-known water-splitting experiment, using external light source on semiconductor surface, was performed in 1972 by Fujishima and Honda [19]. A UV light photo-active anode was used to split water into oxygen-generating protons traveling through the solution to reach the cathode surface. There protonic reduction occurred and hydrogen was produced. This approach was a tremendous discovery that opened many perspectives on further studies about ‘photo-electrochemistry’ or ‘synthetic photo-synthesis’ [20, 21]. However, the photo-anode efficiency should still be optimized and enhanced to fill in the major part of the solar spectrum. The semiconductor used in this study was TiO2 whose behavior is well described in the following theoretical part.
In a previous work by our group aiming at developing anode materials for solar water-splitting systems [22], TiO2 thin layers with competitive microstructural and photocatalytic properties have been prepared using a one-step low-frequency plasma-enhanced chemical vapor deposition (PECVD) process, operating at temperature as low as 350 °C. Single-oriented pure and N-doped anatase films have been synthesized using titanium isopropoxide (TTIP) as a precursor, and NH3 as a doping agent. Optimized PECVD conditions have enabled to obtain homogeneous micro-columnar porous thin films with thicknesses close to 500 nm. Depth-profiling XPS analyses have proved the nitrogen incorporation into TiO2 lattice after ammonia introduction in the deposition chamber. As another proof of N-doping, Raman and XRD peaks shifting have been observed. Such thin films have been demonstrated to be efficient photocatalytic materials whose activity region can be tailored from UV to visible region by adjusting the proportion of doping agent in the plasma phase.
The present work is a sequel to the optimized TiO2 (non-doped) and N-TiO2 (nitrogen doped) low-frequency PECVD thin films previously developed [22]. The structural, optical, and photo-electrochemical properties of slightly doped (implementing 5 Pa as NH3 partial pressure in the PECVD chamber) layers are investigated in the dark, under UV, and under visible light and compared to the non-doped TiO2 synthesized at two different plasma discharge powers.
2 State-of-art on the photo-electrochemical behavior of semiconductors
The overall water-splitting reaction is summarized as follows: H2O → H2 + ½ O2. For ideal electrolysis systems, for example, those using highly conductive metals as electrodes, a half mole of O2 and a mole of H2 are produced per mole of water. However, for systems using semiconductors or organic-based compounds, yield limitations generally appear due to the use of a low conducting electrode [23,24,25]. Still, water electrolysis requires an external electrical power source in order to drive the reactions.
One interesting approach for water splitting, derived from electrolysis, is photo-electrolysis of water. In such a process, water dissociation into its own elements is induced by solar energy on a single photo-electrode surface. This device eliminates the need for a separate power generator, reducing overall costs and increasing efficiency. The usual suggested hypothesis was the possibility of ‘electrodeless’ water photo-splitting. Cohen explained that water is completely transparent to a certain light range and could never be directly dissociated under energy sources of wavelengths higher than 190 nm. This means that electrode materials, properly responding to common light sources, are required [26]. Semiconductors are widely known for their reaction abilities after absorbing a light source of wavelength smaller than their band gap, i.e., the energy difference between the filled valence band (VB) and the empty conduction band (CB).
When the material absorbs radiations of wavelengths at least corresponding to their band gap energy (according to the formula: Eg = hc/λ, Eg is the band gap energy in eV, h is the Planck constant (6.62·10−34 J s), c is the light velocity (3·108 m s−1), and λ is the wavelength in nm; reductive electrons (e−) are promoted from the Highest Occupied Molecular Orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO). This generates oxidizing holes (h+) in the valence band leading to electron/hole pairs. The limiting photoactivity factor is the lifetime of the electron–hole pairs. Increasing this latter factor leads to a higher material efficiency under light because of enhancement of redox reactions rate. According to literature [27, 28], semiconductors have two different behaviors (p-type or n-type) in contact with a solution depending on the energy edges depletion on the material surface (Fig. 1).

Schematic representation of intrinsic energy band behaviors of a n-type semiconductor and b p-type semiconductor
An n-type semiconductor (Fig. 1a) has an electron excess near the conduction band. The level depletion derivative force acts in such a way to transport electrons into the electronic back contact through the external circuit towards the facing counter-electrode (reduction), while holes are preferably emitted into the solution. These holes are able to oxidize species in the electrolyte and the semiconductor behaves as a ‘photo-anode.’ A p-type semiconductor (Fig. 1b) has a hole excess near the valence band that are transported into the back contact by the derivative force while electrons pass through the solution where they can reduce species in the electrolyte. Thus, such a semiconductor is qualified of a ‘photo-cathode.’ These behaviors are observed on both intrinsic (non-modified by external species) and extrinsic (modified by metals or chemically doped) semiconductors. The latter explanations are important to understand the electrochemical systems proposed and studied in the literature.
The present work is mainly focused on a configuration in which a light activated photo-anode oxidizes water into oxygen (H2O + 2h+ → 2H+ + ½ O2). The generated protons diffuse through the H2SO4 electrolyte while the electrons flow through the external circuit. The protons reduction into hydrogen occurs at the glassy carbon electrode surface (2H+ + 2e− → H2). The present work is more a study in the field of photo-water splitting or photo-electrochemical water molecule dissociation.
The choice of semiconductors as electrodes is not random [29]. These materials should have enough large band gaps to split the water molecule into its own elements hydrogen and oxygen. Numerous studies on photo-water splitting have been developed on common semiconductors. The most important point is that the semiconductor band gap edges must fill in the water oxidation/reduction levels which are theoretically 1.23 eV per electron (oxidation) and 0 eV (reduction) (vs. RHE, reversible hydrogen electrode). This is the reason why silicon is limited for hydrogen generation from water even though its band gap could cover a major part of the solar spectrum (1.1 eV–2.4 eV).
Here, a remark should be done considering that confusions occur generally between three concepts in the ‘photo-active materials’ field. The three concepts are ‘photovoltaic,’ ‘photo-catalysis,’ and ‘photo-electrochemistry.’ In fact, ‘photovoltaic’ process is converting solar light into electricity. This concept is applied to Solar Cells (SC) manufacture and elaboration [30, 31].
‘Photo-catalysis’ is more like accelerating a reaction under light effect. Photocatalytic effect is actually applied for organic molecules degradation and water purification using economical activation sources such as natural light [32,33,34].
‘Photo-electrochemistry’ or ‘Photo-galvanic’ process is the study of complex chemical reactions induced or enhanced by light irradiation [35]. This is the field that interests our study in this paper.
Figure 2 is a schematic representation of possible semiconducting materials with their band edges for water-splitting application (from reference [36]). For hydrogen generation by water splitting, a compromise should be made between a gap energy corresponding to the required light wavelengths domain (ultraviolet, visible, and infrared) and band gap edges well positioned according to the water oxidation/proton reduction redox couples. Taking the example of ZrO2 band edges, it could easily oxidize water into oxygen and induce hydrogen generation only when irradiated with short wavelengths (for example, 5 eV: λ ≤ 250 nm) situated in the UV range. In the case of silicon (Si), the wavelength absorption domain is enlarged (λ ≤ 1130 nm) but this material could not be used to produce hydrogen by water splitting due to its valence band edge situated above the O2/H2O oxidation energy level.

Band gap energy edge position according to water oxidation/proton reduction redox couples for thirteen common semiconductors [36]
In the present work, TiO2 has been used as a photo-anode semiconductor. As it can be noticed in Fig. 2, titanium dioxide presents good valence band position relatively to water oxidation redox couple and it has an average gap value (λ ≤ 414 nm).
3 Experimental section
3.1 Pure and Nitrogen-doped anodic layers synthesis procedure
Both TiO2 and N-TiO2 layers were deposited by a low-frequency PECVD process according to the procedure previously described and optimized in a paper by our group [22]. The TiO2 chemical precursor is Titanium Tetra-isopropoxide TTIP (purity ≥ 97%) from Sigma Aldrich. The doping agent is anhydrous ammonia NH3 supplied by a gas cylinder from Gazechim®. The partial pressures of vapors and gases in the PECVD chamber during the deposition step are 22.5 Pa for Ar + TTIP (Ar being used as a carrier gas), 17 Pa for O2 (O2 being used as an oxidizing agent), and 5 Pa for NH3 (added in the plasma reactor for doping purpose). Two sample series were prepared: the series at low plasma discharge power (64 W) and the series at high plasma discharge power (100 W). 64 W has been chosen as the same low input power as that optimized in a previous study by our group dedicated to TiO2 deposition by radio-frequency PECVD [37] and 100 W has been selected as the highest input power delivered by the low-frequency generator used in this study. The deposition time was 20 min and the deposition temperature was 350 °C for all the samples, in a one-step process.
In this paper, TiO2 and N–TiO2 films prepared at 64 W and 100 W will be named 64W–TiO2, 64W–N–TiO2, 100W–TiO2, and 100W–N–TiO2, respectively.
Three different kinds of supports were used: single crystalline silicon (100) (from the Monsanto Electronic Materials Company, Boron-doped type p-Si (100), resistivity: 1–50 Ω), nickel foil (purity ≥ 99.9%) for structural characterizations, and a porous carbon substrate (commercial E-Tek® conductive black cloth for electrochemical characterizations).
3.2 Morphological and structural characterization techniques
In the previous work by our group [22], the morphology of plasma layers had been observed for materials deposited on Si substrates. Here, the surfaces and cross sections morphologies were investigated on layers deposited on Ni (for crystal morphology observation) and E-Tek® (for electrochemical functional studies) supports by scanning electron microscopy (SEM) using a Hitachi S4800 microscope.
The density of plasma layers deposited on Si was determined by X-ray reflectometry (XRR) using a Siemens/Bruker D5000 with a CuKαsource (λ = 1.54056 Å). Manipulating with the same Si support, ESCALAB 250 from Thermo Electron was used for XPS depth profile correlations and a GES5E spectroscopic ellipsometer from Semilab (wavelength from 250 nm to 1000 nm, incidence angle of 70°) was used to evaluate the layer refractive index and optical properties. The ellipsometry data were simulated and fitted using Winelli2® software.
3.3 Electrochemical characterizations in a single compartment photo-electrochemical cell
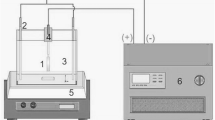
The electrochemical characterizations of plasma-layered electrodes were performed in a single compartment Teflon® cell equipped with a 27-mm diameter transparent quartz window (Fig. 3). A three electrode configuration was implemented involving: a plasma layer deposited on an E-Tek® carbon cloth (in the form of a disc of 10 mm diameter making a constant surface of 0.785 cm2) as the photo-anode (working electrode), a commercial glassy carbon material (0.7 cm x 2 cm) as the cathode (counter-electrode), and a commercial Ag/AgCl (3 M KCl) from SI Analytics® (0.197 V vs. RHE) as the reference electrode.

Schematic representation of the single compartment photo-electrochemical cell
The three electrodes were immersed in a 0.5 M H2SO4 liquid electrolyte solution (prepared from a 100% H2SO4 solution from Merck Millipore® in 18 MΩ pure MilliQ® water from Millipore®). In order to ensure good electron collecting through the external circuit, both photo-anode and cathode were tightly connected to gold wires (Goodfellow ≥ 99.9% from Sigma Aldrich®) that were not immersed in the electrolyte solution in order to avoid metallic interference response. Two different irradiation sources were used: a 75 W (λ = 355 nm) [37] from Eurosolar® and a 150 W filtered (λ ≥ 400 nm) from Van Cliff lamps for UV and visible, respectively. It should be mentioned that since the carbon cloth is not transparent and by that it will absorb the light before reaching the active layer, the plasma film was facing the lamp irradiation which was focused and adjusted to the cell quartz window in order to maintain constant irradiation in UV and visible for all the samples.
The whole cell was covered by aluminum foil during the illumination experiments to concentrate the light and to avoid room light interference when performing experiments in the dark. Lastly, before each electrochemical test, the electrolyte solution was degassed by pure argon bubbling for 40 min in order to get rid of the dissolved oxygen.
Impedance measurements were performed in order to evaluate the TiO2 and the N-TiO2 intrinsic electronic resistance and the electrode/electrolyte interface properties. Electrochemical Impedance Spectroscopy (EIS) was performed with a Solartron® SI 1260 frequency analyzer (in a range of 0.02 Hz–105 Hz with 10 mV AC amplitude). Both ZPlot® and ZView® softwares were used to record and fit the experimental data.
In order to investigate the n-type semiconductor behavior and the Oxygen Evolution Reaction (OER), the Open Circuit Voltage (OCV), the Cyclic Voltammetry (CV) at 5 mV s−1, and the Linear Voltammetry (LV) at 1 mV s−1 were, respectively, recorded using a Solartron® SI 1287 galvanostat–potentiostat. The softwares are CorrWare 2® for experimental data recording and CorrView® for text files collecting.
4 Results and discussions
4.1 Coatings morphology and thickness observations by SEM
In a previous paper by our group [22], SEM characterization of films on Si substrate had enabled to observe micro-columnar porous thin films with thicknesses close to 500 nm. Nevertheless, the crystal morphology could not be precisely depicted. This is the reason why dense nickel foils were used as supports in the present study (Fig. 4).

Surface and cross section SEM images of a 64W–TiO2, b 100W–TiO2, c 64W–N–TiO2, and d 100W–N–TiO2 deposited on nickel foil
Figure 4a shows the 64W–TiO2 sample on Ni, the (101) facet orientation is clearly observed. Moreover, a dense thin layer starts to grow at the bottom of the upper layer for such a plasma power, while the 100W–TiO2 layer (Fig. 4b) shows a homogeneous columnar growth throughout the thickness. The latter layers expose ‘flower-like’ (101) facets while sharper and defined ones are exposed for the 64W–TiO2 layers. In Fig. 4c, d, respectively, corresponding to the 64W–N–TiO2 and 100W–N–TiO2 doped samples, the (101) facets coverage with a nitride shell is obviously observed as cited in the works of Cho et al. [38].
The coverage is more observed in the case of layers prepared at 64 W since the exposed facets are dense unlike the ‘flower-like’ facets of the layers synthesized at 100 W where the nitrided shell could have covered the scattered surface.
The TiO2 and N–TiO2 low-frequency PECVD thin layers deposited on porous carbon (E-Tek®) are presented in Fig. 5. The ‘flower-like’ morphology of the crystallized coatings depicted on Ni support could be noticed on the carbon cloth surface. The nitrogen doping seems to induce local agglomeration on some surface points. The crystallite edges become a little thicker after nitration.

Surface and cross section SEM images of a 64W–TiO2, b 100W–TiO2, c 64W–N–TiO2, and d 100W–N–TiO2 deposited on an E-Tek® carbon cloth
Table 1 presents the films thicknesses measured by SEM cross section on nickel foil and E-Tek® cloth for the same deposition time of 20 min.
The first observation is that increasing the plasma power from 64 W to 100 W increases the growth rate since higher thicknesses were measured on both substrates at 100 W. The second observation is that lower thicknesses values were obtained on E-Tek® cloth. This could be due either to a hidden thickness by crystals grown inside the cloth or to the fact that metallic nucleation sites on nickel foil are highly activated by plasma and accelerated by the layer surface growth. The third observation is that the growth rate is slowed by doping process, especially on Ni support. In fact, when adding NH3 gas into the plasma phase, reactive species reactions are more favored into the plasma gaseous volume more than on substrate surface. This leads to a competition between species–species (volume) collisions and species-solid (substrate) interactions [39]. This process leads to lower growth rate.
4.2 Coatings refractive index, growth simulation, and Nitrogen composition
The layers refractive index and growth simulation were analyzed by ellipsometry measurements on Si wafer. This support is the most suitable for this kind of analysis since it is dense and reflective so the layer–substrate interface could be easily detected by the incident light beam.
According to the literature, the theoretical refractive index for crystalline dense anatase is 2.5–2.6 [40, 41]. The RI of TiO2 thin films could be affected by the layer microstructure including lattice modification, porosity, and crystalline amount [42].
In our case, the layer was analyzed at atmospheric pressure and its microstructure was fitted on the wavelength range 380 nm–1 µm using the stack model presented in Fig. 6.

TiO2 and N-TiO2 layer Cauchy simulation by ellipsometry on Si (100) wafer
The Cauchy optical behavior of dielectric materials well fits the bulk TiO2 material on this range. A diffusion layer, based on the Bruggeman effective medium approximation of the underlying bulk TiO2 mixed with the upper air, was used to simulate the top surface roughness of the films which could play a significant role in the interface processes in contact with the electrolyte.
Table 2 shows the layer microstructure composition with the corresponding refractive index given at 633 nm (RI).
It can be noticed that all the correlation factors are above 0.986 which is an indication of a good layer microstructure fitting with the Cauchy behavior and the chosen model of Fig. 6. As a microstructure correlation with XPS atomic percentages, the atmospheric nitrogen adsorbed atomic percentages have been found to be 0.55 at% at 400.14 eV on the non-doped 64 W and 0.79 at% at 399.93 eV on the non-doped 100 W layers. This adsorbed nitrogen is generally located in the diffusion layer. For almost the same diffusion percentages at non-doped 64 W (5%) and 100 W (4%), the RI lowering from 1.9794 (64 W) to 1.5937 (100 W) is mainly due to the disorder induced by N2 surface adsorption. Meanwhile, the non-doped TiO2 bulk keeps almost a constant RI for both powers as an indication of lattice conservation. This RI is lower than the theoretical anatase one even though the layer has been proved to be well crystallized at 350 °C [22]. This fact is probably due to the layer porosity (nair = 1 < nanatase, where n is the refractive index of the medium) so that a porous layer introduces more air since the analysis is performed under atmospheric surrounding. This will be discussed in the XRR analysis.
For the N-doped TiO2 samples at 64 W and 100 W, the optical correlation with the XPS results is deduced from Table 3.
The percentage values in Table 3 represent the mean value of the N1s atomic contents along the thin film from the surface (0 s etching time) to 2110s etching time. Both N-doped 64 W and 100 W layers exhibit mostly the same insertion and adsorbed nitrogen atomic percentages, so this is not the limiting factor in that case. The difference is observed in the substitutional (Ti-N) nitrogen amount. As a correlation with the RI values, it is obvious that increasing the substitutional N-doping induces a bulk RI decrease (1.9219 for 64 W and 2.4693 for 100 W).
Even though the insertion and surface adsorption percentages are similar in both plasma powers, the diffusion RI decrease from 1.8803 (64 W) to 1.6205 (100 W) is explained by the higher diffusion concentration at 100 W.
These microstructure differences between two layers synthesized at two different plasma powers are responsible of the behavior difference at the electrode–electrolyte interface presented in the electrochemical results.
4.3 Coatings density
The theoretical density reported for anatase is 3.82–3.97 g.cm−3 [43]. Using XRR data, a thin layer mass density ρm is calculated according to the formula 1 [44]:
\({\Theta _{\text{c}}}\) is the critical angle (converted from degrees to radians) deduced from the minimum of the first derivative of the intensity versus angle plot, λ = 1.54056 Å (of the incident beam), NA is the Avogadro number (6.022·1023 e.mol−1), r0 = 0.0000282 Å is the standard electron radius, Ai is the element molar mass, Ci is the element atomicity, and Zi is the element atomic number.
The first derivative plots as a function of the angle for the non-doped and the doped 64 W and 100 W layers on Si (100) are plotted in Fig. 7.

XRR first derivative plot of the non-doped and N-doped 64 W and 100 W layers deposited on reflective Si (100)
For the non-doped 64 W and 100 W layers, \({\Theta _{\text{c}}}\) were found to be 0.25579° and 0.2285° (the mean value of the two minima on the curve), respectively. Using the formula cited previously, the calculated 64W–TiO2 density is 3.27 g cm−3 and the calculated 100W–TiO2 density is 2.61 g cm−3. The two values are lower than the theoretical 3.82–3.97 g cm−3 as an indication of porosity existence. The 100W–TiO2 exhibits more porosity than the 64W–TiO2 which is in good agreement with the RI values. This could also be linked to the SEM observations shown in Fig. 4a and b where the 100W–TiO2 surface presents a ‘flower-like’ porous morphology that has a low RI of 1.5937 due to atmosphere inclusion.
The N-doped TiO2 molecular formula is TiO2−xNx since some Oxygen atoms are substituted by Nitrogen. According to that, the multiplying factor ∑AiCi/∑ZiCi would be equal to the following:
The critical angles were found to be 0.24761° and 0.26156°, respectively, for 64W–N–TiO2 and 100W–N–TiO2. Not considering the multiplying factor, the calculated values of the coating density are 1.46 mol cm−3 and 1.62 mol cm−3, respectively, for 64W–N–TiO2 and 100W–N–TiO2. Then, the multiplying factor converts the unit from mol cm−3 to g.cm−3.
The multiplying factor development leads to the following simplified ratio: (112 − 18x)/(80 − 9x). According to XPS results, x64 > x100 (x is the number of substituted oxygen, 64 and 100 refer to the plasma powers of 64 W and 100 W) since more oxygen atoms are substituted at 64 W. So:
Following this mathematical procedure, the multiplying factor at 64 W is lower than the one at 100 W. This result leads to a higher density for the 100W–N–TiO2. This is also in good agreement with the optical RI values that are higher at 100 W (doped) than at 64 W (doped) as an experimental proof of the higher lattice modification at low plasma power.
In fact, according to some references [45, 46], the variation in the nitrogen bulk amount is plasma power dependent. This is due to the nitrogen form present in the gaseous phase and which is directly related to the electronic density (the plasma power). The NH3 dissociation reaction in plasma phase has been well reported in reference [46]. It has been demonstrated that, at the same chamber total pressure (which is the case for both doped samples), NH3 requires little plasma power to dissociate. Thus, the N+ species intensity is higher in the low plasma power ranges and then reaches an almost steady state where N+ species intensity increases slightly up to 150 W. This explains why the substitutional N-doping is more pronounced at 64 W and leads to optical and microstructural modifications. At 100 W, the bulk microstructure is less affected by nitrogen substitution while the surface state modification seems to be more expressed.
4.4 n-Type semiconducting behavior by OCV investigation
The definition of OCV of a photo-electrochemical system is the equilibrium potential without applying any external current. According to reference [47], when the semiconductor surface is in contact with the liquid electrolyte, a junction is formed. The latter one, for a n-type semiconductor surface, is represented in Fig. 8.

Electrode surface-electrolyte junction a real photo-electrochemistry and b simulated at Open Circuit Potential for a n-type semiconductor schematic representations
In real photo-electrochemistry conditions (Fig. 8a), and in case of a n-type semiconductor (like reported for TiO2), the light irradiation induces electron–hole separation and the added external bias voltage plays the role of the derivative force that conducts the electrons through the external circuit towards the cathode (reduction site) and the holes to the anode surface (electrolyte molecules oxidation).
At OCV conditions (equilibrium state where no current is added in Fig. 8b), when the supposed n-type semiconductor is irradiated, electron–hole separation occurs. Since there is no derivative force to conduct the electrons through the external circuit, these species in the depletion region migrate to the electrode surface and from there to the electrolyte [48]. Thus, under irradiation, the n-type semiconductor Fermi level shifts to lower energy levels towards the electrolyte redox potential. According to reference [49], the OCV (defined as an electrical field) is given by the following equation:
where e is the elementary charge, Voc is the OCV, Ei(Fermi) is the Fermi energy level, and Eeq is the maximum level lowering when the anode is in contact with the electrolyte.
When the light is switched on, EFermi gets closer to Eelectrolyte and Voc drops (since e, the elementary charge, is a constant). When the light is switched off, a relaxation phenomenon occurs and the Voc increases again.
The OCV of the pure and doped TiO2 layers were evaluated under UV and visible irradiation for 800 s. The results are presented in Fig. 9.

OCV measurements under UV and visible light for a 64W–TiO2, b 100W–TiO2, c 64W–N–TiO2, and d 100W–N–TiO2 deposited on E-Tek®
It is clear that the four layers exhibit a n-type semiconducting behavior since the OCV increases when the light is switched off and decreases again under illumination.
For the pure 64 W and 100 W TiO2 layers (Fig. 9a, b), the Voc is lower in the UV light than in the visible light. This is an indication that the Fermi level of the materials is closer to the electrolyte redox potential in UV. This conclusion is logical since the non-doped TiO2 absorbs the radiations at least corresponding to its gap in the UV.
The initial Voc decrease during the first 100 s of illumination is another proof of UV light absorbance. However, the Voc remains mostly stable under visible light as an indication that the Fermi level is not affected under visible light illumination for pure samples.
The remarkable point here is that the Voc drop when switching on the UV light again at 500 s is fast and reaches a steady state at 700 s for the 100W–TiO2 while it is slower and is still decreasing at 800 s for the 64W–TiO2. This means that the electron–hole pairs are instantly generated under UV illumination in the case of 100 W–TiO2.
The Voc reversibility under visible light has to be discussed for the 64 W and 100 W pure TiO2 (Fig. 9a, b, light gray plots). When the lamp is switched on at 500 s, the Voc drops quickly to its initial value in the case of 64 W (reversible drop) while it decreases slowly and to a higher Voc value in the case of 100 W. This phenomenon is explained in Fig. 10.

OCV schematic variation under illumination for 100W–TiO2
Correlating the Voc variations to the surface morphology and optical properties as determined in Sect. 4.2, some surface electron trap seems to exist in the 100W–TiO2. In fact, several experimental observations prove this hypothesis. The atmospheric N2 adsorption is higher at 100 W (0.79%) than at 64 W (0.55%) as reported in XPS analysis. Moreover, the adsorbed N at 100 W is located at a lower binding energy (399.93 eV) than at 64 W (400.14 eV). In addition to that, the ‘flower-like’ surface observed in the case of 100W–TiO2 seems to play a significant role in the material behavior at the electrode–electrolyte interface. The conclusion from Fig. 10 for the visible light non-reversibility of the Voc for 100W–TiO2 is that, under the first 100 s irradiation, the value is mainly stable. Switching off the light induces some state relaxation but when the light is switched on again, some electrons, still trapped in the surface state from the first 100 s illumination, limit the Voc decrease to its initial value.
Similar explanations could be presented in the case of the nitrogen-doped TiO2 layers (Fig. 9c, d). The Voc initial values under visible light are lower than under UV as a proof of the doping role in the charge separation enhancement in the visible range. Nitrogen doping creates N1s trap levels which leads to a decrease of the gap energy. Thus, the initial Fermi level is closer to the electrolyte redox potential and the Voc shifts to lower values. More N1s levels are generated in the band gap of the doped 64 W which lowers the Voc in both UV and visible light compared to the same layer synthesized at 100 W. These levels also play the role of electron traps so that it could be seen that the Voc stabilizes better in the case of the 64 W layers.
4.5 Electrode–electrolyte interface by EIS and correlation with I–V curves by CV
The electrode–electrolyte interface has been studied in the dark, in UV and in visible light by EIS in the conditions previously mentioned in Sect. 3.3. The results are presented in Fig. 11.

EIS spectra at 0 V vs. the Ag/AgCl reference electrode of the 64 W and 100 W layers on porous carbon in the dark, in UV and in visible light for the a non-doped TiO2 layers and b N–TiO2 layers
The decrease of the charge transfer resistance (Rct) by the doping process is obvious by the scale lowering from the non-doped to the doped samples.
It could be noticed that no Rct modification occurred under visible light for the non-doped samples at both 64 W and 100 W. This is an indication that the charge transfer at the electrode–electrolyte is not affected by this wavelength range (> 400 nm) which is logical due to the band gap of both materials. In addition, the resistances of the layers deposited at 100 W are slightly lower than those of the films prepared at 64 W revealing a more intrinsic conductive microstructure at higher plasma power. Under UV light, the resistances decrease remarkably in the case of both plasma powers. This result is in good agreement with both materials band gap and is also coherent with the previous observation since the Rct at 100 W decreases more than at 64 W revealing another time the 100 W layer more conductive intrinsic properties.
For the 64W–N–TiO2, it could be observed that Rct decreases remarkably from the dark to UV. A good decrease is also observed under visible light as an indication of charge separation and mobility under this wavelength range (> 400 nm). In parallel, for 100 W–N–TiO2, the Rct decrease under UV light is almost not observed while it decreases under visible light. This means that the charge separation and mobility are better under visible light than under UV light.
The explanation of such differences is reported in the anodic CV curves (at slow scan rate of 5 mV s− 1) and presented in Fig. 12.

CV curves at 5 mV.s− 1 in the dark, in UV, and in visible light for a 64W–TiO2, b 100W–TiO2, c 64W–N–TiO2, and d 100W–N–TiO2
Figure 12a, c reveals obviously the semiconducting behavior of the 64 W non-doped and N-doped TiO2 on porous carbon. In the dark, no current was measured for these two layers. In the case of the 64 W–TiO2 layer (Fig. 12a), under UV light, the photo-current remarkably increased and the redox reaction peaks (anodic Ipa and cathodic Ipc) are displayed. The positive peak is attributed to the oxidation of Ti3+ appearing under UV illumination (Ti3+→Ti4++1e−) while the negative peak corresponds to the reduction of the generated Ti4+ species (Ti4++1e−→Ti3+) [50]. Moreover, both peaks (anodic Epa and cathodic Epc around 0.35 V–0.4 V) are close enough to consider a fast charge transfer which has been observed in EIS spectra under UV for this layer (Fig. 11). Under visible light, no photo-current was detected and no charge transfer occurred which is in good agreement with EIS results shown in Fig. 11. In the case of the 64W-N-TiO2 layer (Fig. 12d), the semiconducting behavior is also obvious under both UV and visible light in agreement with the EIS observations. Moreover, the generated photo-current is greater than the one in the non-doped 64 W TiO2. This shows the relevant advantage of nitrogen doping in TiO2. Epc slightly shifts to the left indicating a faster charge transfer under visible light. The conclusion is that the layers prepared at 64 W show a typical n-type semiconducting behavior and could be qualified of ‘typical photo-materials’ which activity is completely light-dependent.
In the case of the 100W–TiO2 layers (Fig. 12b), the Ti3+ [51, 52] species already exist in the material without illumination. The proof is the oxidation and reduction peaks existing in the dark. Under UV light, the photo-current increases especially in the 0–0.2 V vs. Ag/AgCl range and the redox peaks intensity increases slightly. This explains the Rct decrease in the UV displayed in Fig. 11. It could also be observed that Rct decreases remarkably from 64W–TiO2 to 100W–TiO2. This fact is due to the greater photo-current generated in the case of 100W–TiO2 due to the species mobility. This mobility results from a combination of the initial Ti3+ and the Ti3+ generated by UV irradiation. Under visible light, the material behavior is almost the same as in the dark proving that the initial Ti3+ does not tailor the layer band gap. The redox peaks intensities are slightly lower than the ones in the dark revealing the surface state role in trapping some mobile Ti3+ decreasing by that the photo-current and increasing the Voc values as reported in Fig. 9 and explained in Fig. 10. For the 100W–N–TiO2 (Fig. 12d), the Ti3+ species still exist intrinsically into the material as the redox peaks exist in the dark. Their concentration increases slightly under UV and visible light as the photo-current increases under irradiation. These results are in good agreement with the material EIS spectra shown in Fig. 11. Still, this increase is not really noticeable which proves the existence of an initial conductive phase for the layers deposited at 100 W. At the interface, in the electrode–electrolyte equilibrium conditions (0 V vs. Ag/AgCl), it could be noticed that a higher photo-current is measured under visible light than in the dark or UV for the 100W–N–TiO2 (see the starting point of the CV curves in Fig. 12d). This reveals the nitrogen role in the band gap tailoring and in the Rct decrease under visible light. Thus, at a higher plasma power, the electrode contains an initial conductive phase including Ti3+. The electrode activity exists even in the dark and is barely enhanced under light (in the UV for the pure TiO2 and in the visible for the N–TiO2). The layers could be qualified of ‘partial photo-materials’ in which carriers are created by light and easily transferred by an initially existing conductive phase.
4.6 LV curves
LV has been performed for the four layers in the dark, UV, and visible light from 0 to 2 V vs. Ag/AgCl at a scan rate of 1 mV s−1. The results are presented in Fig. 13.

LV curves at 1 mV.s−1 in the dark, UV, and visible for a 64W–TiO2, b 100W–TiO2, c 64W–N–TiO2, and d 100W–N–TiO2
Linear Voltammetry is a useful technique to study the oxygen evolution reaction (OER) current and the starting potential.
The photo-current is enhanced under UV light for the 64W–TiO2 as observed in Fig. 13a. This result is in good agreement with the previous electrochemical observations. In the case of the 100W–TiO2 (Fig. 13b), no current variation is observed between dark and visible light. The currents are higher than in the case of 64W–TiO2 as an evidence of initial conductive carriers in the material. These carriers are highly activated under UV light, their concentration increases under this light range and since the layer is initially conductive, they migrate to the electrode–electrolyte surface causing instant electrolyte oxidation (at 1.4 V vs. Ag/AgCl). At the same time, an amount of Ti3+ could be oxidized to Ti4+ which causes current decrease and then lowering at 2 V vs. Ag/AgCl [51, 52]. Similarly, the photo-current enhancement under visible light for the 64W–N–TiO2 (Fig. 13c) and 100W–N–TiO2 (Fig. 13d) is clear.
The higher current observed in the case of the 100W–N–TiO2 reveals the more intrinsic conductive properties due to the initial Ti3+ species.
5 Conclusions
Anatase and Nitrogen-doped anatase thin layers with thicknesses of some hundred nanometers have been grown by low-frequency PECVD process at 64 W and 100 W according to previously optimized conditions. Three different substrates have been used for the deposition of layers: silicon for XRR, XPS, and ellipsometry measurements, Nickel foil for crystal growth morphology and nitride shell observations by SEM, and conductive porous carbon (E-Tek®) for electrochemical studies. Due to its intrinsic porosity, lower layer thicknesses have been obtained on the porous carbon when compared to dense substrates. XPS has revealed a higher substitutional Ti-N incorporation into the doped layers prepared at 64 W due to the higher amount of N+ species in the lower plasma power ranges as previously reported in the literature. Ellipsometry microstructure simulation has shown that the films are rough, constituted of a bulk TiO2 on top of which lies a diffusion layer. In parallel, the layer refractive index variation from the theoretical value for anatase (2.5) was more noticed at 64 W than at 100 W. This is an indication of a higher lattice modification at lower plasma power as shown by XPS. These structural differences directly related to the plasma power in the preparation of films have a direct impact on the material photo-electrochemical behavior where the layers prepared at 64 W exhibit a ‘fully semiconducting’ behavior under UV and visible light, whereas the films deposited at 100 W already contain a conductive phase (Ti3+ species) which activity can be enhanced under light.
As perspectives, layers with higher nitrogen amount should be characterized under the same conditions in order to evaluate the effect of doping incorporation on the structural, optical, and photo-electrochemical properties of anatase PECVD materials. Then optimized N-doped films should be integrated as anodes in photo-electrochemical systems for hydrogen generation by water splitting under solar light.
References
Ribeiro H (2002) Fossil fuel energy impacts on health. Encycl Life Support Syst 1–9
Princen T, Manno J, Martin P (2015) The fossil fuel problem. In: Ending of fossil fuels era, pp 3–97
Holtsmark B (2015) A comparison of the global warming effects of wood fuels and fossil fuels taking albedo into account. GCB Bioenergy 7:984–997. https://doi.org/10.1111/gcbb.12200
Scientists U of C (2016) The hidden costs of fossil fuels. In: Science for a healthy planet and safer world
Chauhan NS, Singh VK (2015) Fundamentals and use of hydrogen as a fuel. J Mech Eng 6:63–68
Jechura J (2015) Hydrogen from natural gas via steam methane reforming (SMR)
Raggio G, Pettinau A, Orsini A et al (2005) Coal gasification pilot plant for hydrogen production. Part A: coal gasification and syngas desulphurization. In: Second international conference on clean coal technology our future
Deiana P, Pettinau A, Tola V (2007) Hydrogen production from coal gasification in updraft gasifier with syngas treatment line
Lensa W, Von, Verfondern K (2010) Coal gasification for hydrogen production using nuclear energy coal gasification for hydrogen production using nuclear energy. In: 18th World hydrogen energy conference, pp 191–198
Lan R, Jin H, Guo L et al (2014) Hydrogen production by catalytic gasification of coal in supercritical water. Energy Fuels 28:6911–6917
Van Niel EW (2016) Biological processes for hydrogen production. Adv Biochem Eng Biotechnol 156:155–193
Orozco-Pulido R (2011) Hydrogen production from biomass by integrating thermo-chemical and biological processes
Loubette N, Junker M, Jacques R, Forbach C (2006) State of the art of biological hydrogen production processes
Das D, Veziroǧlu TN (2001) Hydrogen production by biological processes: a survey of literature. Int J Hydrogen Energy 26:13–28
Carmo M, Fritz DL, Mergel J, Stolten D (2013) A comprehensive review on PEM water electrolysis. Int J Hydrogen Energy 38:4901–4934
Kasai S (2014) Hydrogen electrical energy storage by high-temperature steam electrolysis for next-millennium energy security. Int J Hydrogen Energy 39:21358–21370
Rashid MM, Al Mesfer MK, Naseem H, Danish M (2015) Hydrogen production by water electrolysis: a review of alkaline water electrolysis, PEM water electrolysis and high temperature water electrolysis. Int J Eng Adv Technol 4:2249–8958
Bhandari R, Trudewind CA, Zapp P (2014) Life cycle assessment of hydrogen production via electrolysis e a review. J Clean Prod 85:151–163. https://doi.org/10.1016/j.jclepro.2013.07.048
Fujishima A, Honda K (1972) Electrochemical photolysis of water at a semiconductor electrode. Nature 238:37–38
Kennedy JH, Frese K (1978) Photo-oxidation of water at a-Fe2O3 electrodes. J Electrochem Soc 125:709–714
Butler MA (1977) Photoelectrolysis and physical properties of the semiconducting electrode WO2. J Appl Phys 48:1914
Youssef L, Kinfack Leoga AJ, Roualdes S et al (2017) Optimization of N-doped TiO2 multifunctional thin layers by low frequency PECVD process. J Eur Ceram Soc 37:5289–5303. https://doi.org/10.1016/j.jeurceramsoc.2017.05.010
Fountaine K, Lewerenz HJ, Atwater H (2016) Efficiency limits for electrochemical water splitting. Nat Commun 7:13706
Duan J, Chen S, Zhao C (2017) Ultrathin metal-organic framework array for efficient electrocatalytic water splitting. Nat Commun 8:15341
Swierk J, Hernandez D, McCool N et al (2015) Metal-free organic sensitizers for use in water splitting dye sensitized photoelectrochemical cells. Proc Natl Acad Sci USA 112:1681–1686
Cohen A (1910) Studien über photochemische Gleichgewichte IV Das Ligchtgleichgewicht Knallgas-Wasserdampf. Berichte der Dtsch Chem Gesellschaft 43:880
Chovet A, Masson P (2007) Cours de physique des semi-conducteurs
Ngô C, Ngô H (2012) Physique des semi-conducteurs
Ma G, Domen K, Hisatomi T (2015) Semiconductors for photocatalytic and photoelectrochemical solar water splitting. In: From molecules to materials. pp 1–56
Würfel P, Würfel U (2016) From basic principles to advanced concepts
McKevoy A, Markvart T, Castaner L (2013) Materials, manufacture and operation. Sol Cells 1–615
Ajmal A, Majeed I, Malik R et al (2014) Principles and mechanisms of photocatalytic dye degradation on TiO2 based photocatalysts: a comparative overview. RSC Adv 4:37003–37026
Tayade RJ, Surolia PK, Kulkarni RG, Jasra RV (2007) Photocatalytic degradation of dyes and organic contaminants in water using nanocrystalline anatase and rutile TiO2. Sci Technol Adv Mater 8:455–462
Khataee AR, Kasiri MB (2010) Photocatalytic degradation of organic dyes in the presence of nanostructured titanium dioxide: Influence of the chemical structure of dyes. J Mol Catal A Chem 238
Grätzel M (2001) Photoelectrochemical cells. Nature 414:338–343
Liu B, Zhao X, Terashima C et al (2014) Thermodynamic and kinetic analysis of heterogeneous photocatalysis for semiconductor systems. Phys Chem Chem Phys 16:8751
Zhou M (2015) Novel photocatalytic TiO2-based porous membranes prepared by plasma-enhanced chemical vapor deposition (PECVD) for organic pollutant degradation in water
Cho HJ, Hwang PG, Jung D (2011) Preparation and photocatalytic activity of nitrogen-doped TiO2 hollow nanospheres. J Phys Chem Solids 72:1462–1466. https://doi.org/10.1016/j.jpcs.2011.08.026
Guimond S, Schütz U, Hanselmann B et al (2011) Influence of gas phase and surface reactions on plasma polymerization. Surf Coatings Technol 205:S447–S450. https://doi.org/10.1016/j.surfcoat.2010.08.131
Nurfani E, Kurniawan R, Muhammady S et al (2016) Effect of Ta concentration on the refractive index of TiO2:Ta studied by spectroscopic ellipsometry. AIP Conf Proc 1725:1–4
Liu H-Y, Hsu Y-L, Su H-Y et al (2018) A comparative study of amorphous, anatase, rutile, and mixed phase TiO2 films by mist chemical vapor deposition and ultraviolet photodetectors applications. IEEE Sens J 1748:1–1. https://doi.org/10.1109/JSEN.2018.2819700
Woo S-H, Hwangbo CK (2006) Optical anisotropy of microstructure-controlled TiO2 films fabricated by glancing-angle deposition (GLAD). J Korean Phys Soc 48:1199–1204
Lazzarelli HN (2010) Blue chart gem identification
Lee A, Van Der (2013) Diffusion, diffraction, réflectométrie et fluorescence de rayons-X à la PAC. In: International year of crystallography, pp 1–35
Chen H, Nambu A, Graciani J et al (2007) Reaction of NH3 with Titania: N-doping of the oxide and TiN formation. J Phys Chem C 111:1366–1372
Swihart MT, Allendorf MD, Meyyapan M (2001) Process control, diagnostics, and modeling in semiconductor manufacturing IV. In: Fundamental gas-phase and surface chemistry of vapor-phase deposition II. pp 1–496
Rahul M (2010) Photoelectrochemical (PEC) and electrical studies on tungsten and molybdenum heteropolyoxometalate thin films
Muhibbullah M, Abdel Haleem A (2015) Estimation of the open circuit voltage of a pn junction based on photo-electrochemical measurements. Trans Mater Res Soc Japan 40:247–252
Muhibbullah M, Golam Mowla Choudhury M, Mominuzzaman SM (2012) An equation of the width of the depletion layer for a step heterojunction. Trans Mater Res Soc Japan 37:405–408
Jie H, Song-Tao W, Xiao-Li C et al (2004) Photoelectrochemical characteristics of nano-titanium dioxide thin films prepared by RF magnetron sputtering. Acta Phys Chim Sin 20:1191–1195
Ji Y, Guo W, Chen H et al (2015) Surface Ti3+/ Ti4+ redox shuttle enhancing photocatalytic H2 production in ultrathin TiO2 nanosheets/CdSe quantum dots. J Phys Chem C 119:27053–27059
Qiu M, Tian Y, Chen Z et al (2016) Synthesis of Ti3+ self-doped TiO2 nanocrystals based on Le Chatelier’s principle and their application in solar light photocatalysis. RSC Adv 78:74376–74383
Acknowledgements
This project has been funded with support from Lebanese University (UL) in collaboration with AZM&SAADÉ foundation. We also thank Arie VAN DER LEE (IEM, Montpellier) for XRR analysis, Didier COT (IEM, Montpellier) for SEM observations, and Valérie FLAUD (ICGM, Montpellier) for thin films profile investigation by XPS.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Youssef, L., Roualdès, S., Bassil, J. et al. Effect of plasma power on the semiconducting behavior of low-frequency PECVD TiO2 and nitrogen-doped TiO2 anodic thin coatings: photo-electrochemical studies in a single compartment cell for hydrogen generation by solar water splitting. J Appl Electrochem 49, 135–150 (2019). https://doi.org/10.1007/s10800-018-1265-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10800-018-1265-4