
Abstract
TiO2 nanotube arrays (NTAs) decorated with controllable Ag particles were prepared by pulse reverse current deposition in AgNO3/NaNO3 aqueous solution, aiming to improve the photoelectrochemical properties of TiO2 NTA electrode in visible-light region. By tuning the pulse current density and deposited charge density, a controllable synthesis of Ag structures was achieved. Excellent photocurrent responses of TiO2 NTAs in UV and visible light regions were achieved by depositing Ag nanorods and nanoparticles, which was attributed to highly efficient charge separation by the Schottky junction at the Ag/TiO2 interface and localized surface plasmon resonance effect of Ag nanostructures.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Owing to the energy crisis and environmental pollution, tremendous efforts have been devoted to utilize solar energy for hydrogen production and organic compound degradation via developing semiconductor-based photocatalysts. TiO2 nanomaterials have been undoubtedly proven to be the most promising photocatalysts due to the high chemical and physical stability, nontoxity and low price [1, 2]. Among various TiO2 architectures, vertically aligned TiO2 nanotube arrays (TNAs) crafted by electrochemical anodization have been extensively studied in view of its merits such as facile synthesis, high-specific surface area, and highly ordered array structure which is favorable for transportation of photo-generated excitions [3,4,5,6,7,8]. Despite the unique properties above, TiO2 NTAs suffer from two major drawbacks: its intrinsic wide band-gap (i.e., 3.2 eV for anatase) leading to the low utilization efficiency of the solar spectrum and its high recombination rate of photo-generated electron-hole pairs. To this end, many strategies have been developed in quest of higher photocatalytic efficiency and utilization of solar light.
Recently, construction of noble metal–semiconductor nanocomposites has emerged as an outstanding approach to improve the photocatalytic efficiency [9,10,11,12]. On one hand, the Schottky junction at the metal/semiconductor interface enhances the separation rate of photo-generated electrons and holes [13,14,15,16]. On the other hand, the surface plasmon resonance (SPR) effect of noble metals (mainly Au and Ag) enables the light adsorption in visible region [12, 17, 18]. Compared with Au, Ag is particularly suitable for industrial applications for its nontoxicity, relatively low price, and easy preparation of Ag–TiO2 composites. Several techniques, such as electrodeposition, photo-reduction, atomic layer deposition, successive ionic layer adsorption, and reaction and solvothermal reduction, have been reported to fabricate Ag–TiO2 NTAs [14, 19,20,21,22,23,24,25,26,27].
As a facile, versatile and inexpensive technique for decorating Ag nanoparticles on TiO2 NTAs, electrodeposition has attracted immense attention. In particular, galvanostatic pulse current deposition (PCD) technique has been recently considered to be highly effective. Compared with the traditional direct current deposition, PCD possesses the advantages in controlling the morphology of deposited particles by tuning the variable process parameters like pulse current density, pulse on-time, and pulse off-time [14, 24]. In our previous studies [14, 21], Ag nanoparticles on the surface of TiO2 nanotubes were synthesized by PCD. However, it is inevitable that some big Ag nanoparticles were always deposited on the top of TiO2 nanotubes, which declined the photocatalytic activity. Therefore, it is highly desired to optimize the electrodeposition parameters for achieving a rational design and effective control of Ag structure. Further, nanoparticle fabrication using PCD with a homogeneous size distribution remains a challenge.
Herein, we demonstrate a facile galvanostatic pulse reverse current deposition (PRCD) approach for tuning the size of Ag nanoparticles on TiO2 NTAs by varying the pulse current density and deposited charge density, respectively. Using reverse anodic pulse is aiming for the removal of the unwanted Ag particles on the top of nanotube arrays via anodic dissolution on the top surface of TiO2 NTAs. Further, the influence of the parameters (i.e., pulse current density and deposited charge density) on the photoelectrochemical properties was systematically explored.
2 Experimental
2.1 Synthesis of TiO2 TNAs
Highly ordered TiO2 TNAs were grown from Ti foil (>99% purity, thickness of 0.1 mm) via anodization using a two-electrode electrochemical cell, where Ti foil served as anode and a Pt foil served as counter electrode (CE). The anodization was carried out at 20 V for 25 min at room temperature without stirring. A glycerol-water (a volume ratio of 2:1) mixed solution containing 0.5 wt% NH4F was used as electrolyte. The as-anodized samples were subsequently annealed in air atmosphere for 2 h at 450 °C to obtain anatase crystallinity.
2.2 Synthesis of Ag–TiO2 TNAs
The Ag–TiO2 NTAs were prepared by the PRCD method using a two-electrode system controlled by an Autolab potentiostat/galvanostat (PGSTAT30). Ti-based TiO2 NTAs with a working area of 1 cm × 1 cm and a Pt foil (2 cm × 2 cm) was used as working electrode (WE) and CE, respectively. The PRCD was carried out at room temperature in an aqueous solution containing 0.01 M AgNO3 and 0.1 M NaNO3 without stirring and the distance between WE and CE was always kept at 1 cm. PRCD consists of cycles of bipolar current pulses with equal amplitude and laxations depicted in Fig. 1. Typically, a 10 ms cathodic pulse with a certain current density was employed for the deposition of metallic Ag and a reverse anodic pulse current with same current amplitude was applied for the preferential dissolution of Ag preventing growth of Ag dendrites on the surface of TiO2 NTAs [28]. Followed by the bipolar pulses, a relaxation of 1000 ms was given to allow the penetration of Ag+ ions inside the channels of nanotubes. The forward duty cycle was determined to be 0.98%. In this study, the influence of deposited charge density and pulse current density was investigated. For the variation of pulse current density, numbers of deposition cycles were adjusted in order to reach the same deposited charge density of 54 mC cm−2 for all the samples. Pulse current densities of 10, 40, 70, and 100 mA cm−2 were employed. For the variation of deposited charge density, the pulse current densities were set as 40 mA cm−2 for all the cases. Deposited charge densities of 18, 54, 180, and 540 mC cm−2 were applied by repeating the deposition cycles (i.e., deposition duration).

The current-time profile of pulse reverse current for electrodeposition of Ag particles (i c cathodic current density, i rev reverse anodic current density, t c cathodic deposition time, t rev anodic dissolution time, t off off time)
2.3 Characterization
The morphology of the obtained samples was examined using a scanning electron microscope (SEM, Hitachi S4800). The crystalline structure of the samples was identified by X-ray diffraction (XRD, Philips, Panalytical X’pert, Cu Kα radiation). UV–vis diffuse reflection spectra (DRS) of the samples were recorded using a Varian Cary-5000 spectrophotometer. The photocurrent measurements were carried out in 0.1 M Na2SO4 using a LHX 150 Xe lamp, a SBP 300 grating spectrometer, and an electrochemical cell with a quartz window. The wavelength-dependent spectral response was measured in a two-electrode configuration with a platinum wire as counter electrode at zero bias in the range of 250–550 nm.
3 Results and discussion
3.1 Effect of pulse current density
Figure 2 presents the representative SEM images of Ag–TiO2 NTAs obtained with pulse current density of 10, 40, 70, and 100 mA cm−2, respectively. It can be seen that the shape of Ag particles varies remarkably with different pulse current densities. Under a pulse current density of 10 mA cm−2, the resulting Ag particles are mainly in micrometer-scaled plate-like shape (Fig. 2a). Notably, most of Ag particles/plates deposit only on the top of TiO2 NTAs under this current density (Fig. 2b). Interestingly, increasing the pulse current density up to 40 mA cm−2, Ag nanorods form and deposit along the inner channel of TiO2 nanotubes (Fig. 2c–d). Similar observation has been reported by Misra and coworkers [29], where TiO2 nanotubes were filled with iron nanorods by pulse current deposition. As the pulse current density further increases to 70 mA cm−2, round-shaped Ag nanoparticles form on the both inner and outer walls and even the bottom of TiO2 nanotubes (Fig. 2e–f). When the pulse current density increases up to 100 mA cm−2, the size of Ag particles becomes even smaller (Fig. 2g–h). The mean diameter and size distribution of Ag particles are investigated by statistical analysis of the particle diameter by measuring 100 particles for each samples (Fig. 3). It is clear that the mean size of Ag particles decreases with the increasing pulse current density (Fig. 3a). Quite intriguingly, the deviation of Ag particles becomes smaller when pulse current density increased, which is further confirmed by the study of size distribution (Fig. 3b). The histograms show that the Ag particles have rather board size distribution under low pulse current density (i.e. 10 mA cm−2) and much narrower size distribution under higher pulse current densities (≥40 mA cm−2) (Fig. 3b). It reveals that a high pulse current density is favorable for the size refinement of Ag nanoparticles. The results of SEM and particle size distribution evidently illustrate that pulse current density strongly influences the morphology and location of the deposited Ag particles on TiO2 NTAs.

SEM images of Ag–TiO2 NTAs obtained under pulse current densities of 10 (a, b), 40 (c, d), 70 (e, f), and 100 mA cm−2 (g, h). A total deposited charge density of 54 mC cm−2 was achieved for each electrode

Effects of pulse current density on size distribution of the diameters of Ag nanoparticles on TiO2NTAs (a) and the corresponding particle size distributions (b). Particle size distributions were obtained by measuring 100 particles from several SEM images
Based on the observation from SEM, it is possible to elucidate the Ag deposition behavior during PRCD. There is a competition between nucleation and cluster growth during the electrodeposition process. Low pulse current density leads to low nucleation rate and fast particle growth rate. Ag+ is mainly consumed at the opening of nanotubes (Fig. 2b). Compared with low pulse current density, moderate pulse current density accelerates the nucleation rate and improves cluster density. Ag+ is consumed inside the nanotube channel. Particle growth is less dominant but remains the major process. Penetration of Ag+ leads to the growth of Ag nanorods along the TiO2 nanotubes (Fig. 2d). High pulse current density causes large over potentials and thus accelerates the nucleation rate and improves the cluster density. Ag+ consumption is mainly for nuclei growth and particle growth is limited. Therefore, highly dispersed nanoparticles can be obtained.
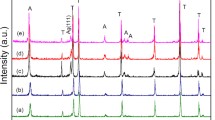
Figure 4 presents the XRD patterns of Ag–TiO2 NTAs obtained with different pulse current densities. The reflections at 25.2° and 48.1° originate from the (101) and (200) planes of anatase structure (JCPDS No. 21-1272). The peaks locate at 38.1°, 44.3°, 64.4°, and 77.7° can be indexed to the (111), (200), (220), and (311) planes of Ag, confirming the presence of metallic Ag in the nanocomposites by PRCD. It is noted that the shape of characteristic peak of Ag (111) phase becomes boarder with the increase of the pulse current density, indicating that the size of Ag particles on TiO2 NTAs decreases. Further, sharp peaks of Ag (200), (220), and (311) phases emerge on Ag–TiO2 NTAs with 10 mA cm−2 and become weaker and boarder with higher pulse current densities. The XRD results further confirm the promoting effect of high pulse current density on the control of small particle size of deposited Ag, which is consistent with the observation from SEM (Fig. 2).

XRD patterns of Ag–TiO2 NTAs obtained under pulse current densities of 10, 40, 70, and 100 mA cm−2. A total deposited charge density of 54 mC cm−2 was achieved for each electrode. A, T and Ag represent anatase TiO2, titanium substrate and metallic silver, respectively
UV–vis DRS was implemented to examine the light absorption property of the Ag–TiO2 NTAs obtained with different pulse current densities (Fig. 5a). The two broad absorption peaks in the spectrum of pristine TiO2 NTAs are attributed to the sub-band gap state of the special tube structure [30]. Intriguingly, all the Ag–TiO2 NTA samples show rather strong absorption in the visible light range of 400–750 nm due to the SPR effect of metallic Ag [12]. The Ag–TiO2 NTAs obtained at 40 mA cm−2 exhibit a maximum red-shift of adsorption edge and strongest absorption in the visible light region, implying that the light absorption was affected by the size of as-deposited Ag particles. In addition, the existing of oxygen vacancies on TiO2 NTAs cannot be ruled out based on our results. However, the anatase TiO2 nanotube arrays annealed in air atmosphere have been reported to only contain negligible amount of oxygen vacancies, while those annealed in oxygen-poor atmosphere like N2 and H2 may be possible to generate oxygen vacancies [31, 32].

UV–vis DRS (a) and photocurrent spectra (b) of Ag–TiO2 NTAs obtained under pulse current densities of 10, 40, 70 and 100 mA cm−2. A total deposited charge density of 54 mC cm−2 was achieved for each electrode
Photocurrent measurement was performed at zero bias potential (vs. SCE) in order to further investigate the effect of particle size by varying the pulse current density on the photoelectrochemical properties of Ag–TiO2 NTAs. Figure 5b presents the photocurrent spectra in the wavelength range of 250–550 nm. In general, a higher photocurrent response indicates a lower recombination rate of the photo-induced electron-hole pairs and a higher transfer efficiency of photo-induced electrons. The spectrum of pristine TiO2 NTAs only shows a maximum peak of photocurrent density (Ip) located at 330 nm, while those of the Ag–TiO2 NTAs display two peaks at 330 and 475 nm, respectively. The poor photocurrent response in the visible light region on the spectrum of pristine TiO2 NTAs indicates that the oxygen vacancies are unlikely to be abundant in pristine TiO2 NTAs. The later peak can be assigned to the SPR effect of metallic Ag particles. With a pulse current density of 10 mA cm−2, Ag–TiO2 NTAs shows a declined photocurrent response in UV region (<400 nm) and negligible SPR effect in visible region (>400 nm), which is very likely due to the light blocking of Ag plates on the top of TiO2 NTA films and less Ag/TiO2 contact. As pulse current densities are higher than or equal to 40 mA cm−2, the photocurrent responses of Ag–TiO2 NTAs are significantly improved in both UV light and visible light regions. The enhanced photocurrent response in UV region can be assigned to the Schottky barriers at the interface of Ag/TiO2 that facilitate the separation of photo-induced electron-hole pairs from TiO2 [14]. On the other hand, under specific visible-light irradiation, metallic Ag can be excited via SPR process to generate electrons, which can be transferred to the conductive band of TiO2 [12]. Similar to the observation in UV–vis DRS, Ag–TiO2 NTAs obtained at 40 mA cm−2 presents a maximum SPR-driven photocurrent response in the visible light region (Fig. 5b). Remarkably, this maximum photocurrent density among is determined to be 24 µA cm−2, which is much higher than reported values from Ag/TiO2 NTAs [14, 21, 33].
There is a balance between particle size of Ag on TiO2 NTAs and photoelectrochemical activity, which has also been often reported in literature [24, 27, 34,35,36,37,38]. In this work, the smallest Ag (~17 nm) uniformly dispersed on TiO2 obtained at the current density of 100 mAcm−2 showed a great improvement in photoelectrochemical activity of TiO2 NTAs, while the medium size Ag nanorods (~59 nm) deposited inside TiO2 nanotubes obtained at the current density of 40 mA cm−2 seemed to be possible to achieve the highest photoelectrochemical activity. Our results are in consistent with a recent study by Valenti and coworker’s study, where they found that small Ag nanoparticles (~15 nm) improved the photocurrent by light absorbing effect, while the large Ag nanoparticles (~65 nm) could further increased the photocurrent by light scattering effect [37]. On the other hand, the photocurrent response measures the photo-generated electrons moving from the perpendicular walls to bottom of TiO2 nanotubes and then to the Ti substrate. However, this electron transfer process is always limited by the poor conductivity of TiO2. We believe that TiO2 nanotubes filled with 1D Ag nanorods could improve the conductivity thus leading to an enhancement on the photocurrent response. Similar strategies have also been reported in the literatures [29, 39].
3.2 Effect of deposited charge density
In another series, the deposited charge density was varied. The cathodic and anodic pulse current densities were set as 40 mA cm−2 for all the samples, according to the finding from Sect. 3.1. Figure 6 presents the representative SEM images of Ag–TiO2 NTAs obtained with deposited charge density of 18, 54, 180, and 540 mC cm−2, respectively. With a deposited charge density of 18 mC cm−2, some Ag nanoparticles are deposited on the opening and the top of TiO2 nanotubes (Fig. 6a). As deposited charge density increased up to 54 mC cm−2, Ag nanoparticles become larger and homogeneously filled inside the TiO2 nanotubes (Fig. 6b). With further increase in deposited charge density to 180 mC cm−2, the size of Ag particles obviously becomes bigger (Fig. 6c). When deposited charge density increases up to 540 mC cm−2, the size of Ag particles grows remarkably up to 2 µm with rather broad size distribution (Fig. 6d). This observation is in good agreement with Faraday’s Law that the amount of deposits is proportional to the quantity of electrical charge. Also, the SEM studies demonstrate that the effect of variation of deposited charge density is more significant on the Ag particle size than on the particle shape.

SEM images of Ag–TiO2 NTAs obtained under electrodeposited charge densities of 18 (a), 54 (b), 180 (c), and 540 mC cm−2 (d). Pulse current densities of 40 mA cm−2 were applied
The XRD patterns of Ag–TiO2 NTAs obtained with different deposited charge densities are shown in Fig. 7, which also confirm the existing of anatase TiO2 and metallic Ag on Ag–TiO2 NTAs. It can been seen that the intensities and FWHMs of characteristic Ag peaks (relative to intensities of TiO2 (101) phase) increase with deposited charge density, implying an increasing trend of particle size.

XRD patterns of Ag–TiO2 NTAs obtained under electrodeposited charge densities of 18, 54, 180, and 540 mC cm−2. Pulse current density of 40 mA cm−2 was applied. A, T and Ag represent anatase TiO2, titanium substrate and metallic silver, respectively
Figure 8a shows the UV–vis DRS of Ag–TiO2 NTAs obtained with different deposited charge densities. Applying a deposited charge density of 18 mC cm−2, the obtained Ag–TiO2 NTA sample shows rather strong absorption in the visible light range of 400–750 nm due to the SPR effect of metallic Ag [12]. Intriguingly, with higher deposited charge density, the obtained Ag–TiO2 NTA samples show near complete light adsorption in the visible light region.

UV–vis DRS (a) and photocurrent spectra (b) of Ag–TiO2 NTAs obtained under electrodeposited charge densities of 18, 54, 180, and 540 mC cm−2. Pulse current density of 40 mA cm−2 was applied
Figure 8b presents the photocurrent spectra of Ag–TiO2 NTAs obtained with different deposited charge densities. With lower deposited charge densities (18 and 54 mC cm−2), the photocurrent responses Ag–TiO2 NTAs are significantly improved in both UV light region (<400 nm) and visible light region (>400 nm). The enhanced photocurrent response in UV region can be assigned to the Schottky barriers at the interface of Ag/TiO2 that facilitate the separation of photo-induced electron-hole pairs from TiO2 [14]. However, Ag–TiO2 NTAs obtained with higher deposited charge densities (180 and 540 mC cm−2) show inhibited photocurrent responses in UV light region and negligible responses in visible light region. It is rational that the small Ag nanoparticles inside TiO2 nanotubes effectively promote the separation rate of photo-induced electrons and holes by the Schottky junction at the Ag/TiO2interface [14], while the huge Ag particles on the top greatly prevent the light adsorption on TiO2 resulting in poor photocurrent responses in both UV and visible-light regions. On the other hand, it also illustrates that photocurrent response are strongly influenced by the Ag particle size. In this work, it demonstrates that Ag nanorods (synthesized under a pulse current density of 40 mA cm−2 and deposited charge density of 40 mC cm−2) other than the small nanoparticles can dramatically enhance the light adsorption and photocurrent response in both UV and visible-light regions.
4 Conclusions
In summary, we have demonstrated that Ag-coupled TiO2 NTAs were successfully synthesized by pulse reverse direct current deposition method. It is found that the Ag particle shape and size were significantly influenced by the pulse current density and deposited charge density. Varying the pulse current density could alter the Ag particle shape and particle size by tuning the nucleation and formation rate of Ag nanoparticles, while varying the deposited charge density could affect Ag particle size. In particular, the high pulse current density can prevent the formation of large Ag nanoparticles at the opening of TiO2 nanotubes, leading to a better penetration of Ag+ ions, and therefore, small Ag nanoparticles uniformly dispersed on the inner and outer surface of TiO2 nanotubes, which showed excellent photoelectrochemical activities (i.e., photocurrent response) in both UV and visible-light regions. Our findings show that pulse reverse current deposition is a promising, facile, and versatile technique to synthesize size-controlled nanoparticles without any additional assistance.
References
Schneider J, Matsuoka M, Takeuchi M, Zhang JL, Horiuchi Y, Anpo M, Bahnemann DW (2014) Understanding TiO2 photocatalysis: mechanisms and materials. Chem Rev 114:9919–9986
Wang MY, Ioccozia J, Sun L, Lin CJ, Lin ZQ (2014) Inorganic-modified semiconductor TiO2 nanotube arrays for photocatalysis. Energ Environ Sci 7:2182–2202
Gong D, Grimes CA, Varghese OK, Hu WC, Singh RS, Chen Z, Dickey EC (2001) Titanium oxide nanotube arrays prepared by anodic oxidation. J Mater Res 16:3331–3334
Beranek R, Hildebrand H, Schmuki P (2003) Self-organized porous titanium oxide prepared in H2SO4/HF electrolytes. Electrochem Solid-State Lett 6:B12–B14
Beranek R, Tsuchiya H, Sugishima T, Macak JM, Taveira L, Fujimoto S, Kisch H, Schmuki P (2005) Enhancement and limits of the photoelectrochemical response from anodic TiO2 nanotubes. Appl Phys Lett 87:243114
Macak JM, Tsuchiya H, Ghicov A, Yasuda K, Hahn R, Bauer S, Schmuki P (2007) TiO2 nanotubes: self-organized electrochemical formation, properties and applications. Curr Opin Solid State Mater Sci 11:3–18
Roy P, Berger S, Schmuki P (2011) TiO2 nanotubes: synthesis and applications. Angew Chem Int Ed 50:2904–2939
Ge M, Li Q, Cao C, Huang J, Li S, Zhang S, Chen Z, Zhang K, Al-Deyab SS, Lai Y (2016) One-dimensional TiO2 nanotube photocatalysts for solar water splitting. Adv Sci. doi:10.1002/advs.201600152
Wang MY, Ye MD, Iocozzia J, Lin CJ, Lin ZQ (2016) Plasmon-mediated solar energy conversion via photocatalysis in noble metal/semiconductor composites. Adv Sci 3:1600024
Jiang RB, Li BX, Fang CH, Wang JF (2014) Metal/semiconductor hybrid nanostructures for plasmon-enhanced applications. Adv Mater 26:5274–5309
Li XH, Zhu JM, Wei BQ (2016) Hybrid nanostructures of metal/two-dimensional nanomaterials for plasmon-enhanced applications. Chem Soc Rev 45:3145–3187
Tian Y, Tatsuma T (2004) Plasmon-induced photoelectrochemistry at metal nanoparticles supported on nanoporous TiO2. Chem Commun. doi:10.1039/B405061D
Ye MD, Gong JJ, Lai YK, Lin CJ, Lin ZQ (2012) High-efficiency photoelectrocatalytic hydrogen generation enabled by palladium quantum dots-sensitized TiO2 nanotube arrays. J Am Chem Soc 134:15720–15723
Xie KP, Sun L, Wang CL, Lai YK, Wang MY, Chen HB, Lin CJ (2010) Photoelectrocatalytic properties of Ag nanoparticles loaded TiO2 nanotube arrays prepared by pulse current deposition. Electrochim Acta 55:7211–7218
Primo A, Corma A, Garcia H (2011) Titania supported gold nanoparticles as photocatalyst. Phys Chem Chem Phys 13:886–910
Nguyen NT, Altomare M, Yoo J, Schmuki P (2015) Efficient photocatalytic H2 evolution: controlled dewetting-dealloying to fabricate site-selective high-activity nanoporous Au particles on highly ordered TiO2 nanotube arrays. Adv Mater 27:3208–3215
Herzing AA, Kiely CJ, Carley AF, Landon P, Hutchings GJ (2008) Identification of active gold nanoclusters on iron oxide supports for CO oxidation. Science 321:1331–1335
Chen MS, Goodman DW (2004) The structure of catalytically active gold on titania. Science 306:252–255
He BL, Dong B, Li HL (2007) Preparation and electrochemical properties of Ag-modified TiO2 nanotube anode material for lithium-ion battery. Electrochem Commun 9:425–430
Roguska A, Kudelski A, Pisarek M, Lewandowska M, Dolata M, Janik-Czachor M (2009) Raman investigations of TiO2 nanotube substrates covered with thin Ag or Cu deposits. J Raman Spectrosc 40:1652–1656
Lai YK, Zhuang HF, Xie KP, Gong DG, Tang YX, Sun L, Lin CJ, Chen Z (2010) Fabrication of uniform Ag/TiO2 nanotube array structures with enhanced photoelectrochemical performance. New J Chem 34:1335–1340
Liang YC, Wang CC, Kei CC, Hsueh YC, Cho WH, Perng TP (2011) Photocatalysis of Ag-loaded TiO2 nanotube arrays formed by atomic layer deposition. J Phys Chem C 115:9498–9502
Wang QY, Yang XC, Liu D, Zhao JF (2012) Fabrication, characterization and photocatalytic properties of Ag nanoparticles modified TiO2 NTs. J Alloys Compd 527:106–111
Lian ZC, Wang WC, Xiao SN, Li X, Cui YY, Zhang DQ, Li GS, Li HX (2015) Plasmonic silver quantum dots coupled with hierarchical TiO2 nanotube arrays photoelectrodes for efficient visible-light photoelectrocatalytic hydrogen evolution. Sci Rep 5:10461
Nishanthi ST, Iyyapushpam S, Sundarakannan B, Subramanian E, Padiyan DP (2015) Plasmonic silver nanoparticles loaded titania nanotube arrays exhibiting enhanced photoelectrochemical and photocatalytic activities. J Power Sources 274:885–893
Zhong JS, Wang QY, Yu YF (2015) Solvothermal preparation of Ag nanoparticles sensitized TiO2 nanotube arrays with enhanced photoelectrochemical performance. J Alloys Compd 620:168–171
Ge MZ, Cao CY, Li SH, Tang YX, Wang LN, Qi N, Huang JY, Zhang KQ, Al-Deyab SS, Lai YK (2016) In situ plasmonic Ag nanoparticle anchored TiO2 nanotube arrays as visible-light-driven photocatalysts for enhanced water splitting. Nanoscale 8:5226–5234
Chandrasekar MS, Pushpavanam M (2008) Pulse and pulse reverse plating—Conceptual, advantages and applications. Electrochim Acta 53:3313–3322
Mohapatra SK, Banerjee S, Misra M (2008) Synthesis of Fe2O3/TiO2 nanorod-nanotube arrays by filling TiO2 nanotubes with Fe. Nanotechnology 19:315601
Lai YK, Sun L, Chen C, Nie CG, Zuo J, Lin CJ (2005) Optical and electrical characterization of TiO2 nanotube arrays on titanium substrate. Appl Surf Sci 252:1101–1106
Mahajan VK, Misra M, Raja KS, Mohapatra SK (2008) Self-organized TiO2 nanotubular arrays for photoelectrochemical hydrogen generation: effect of crystallization and defect structures. J Phys D Appl Phys 41:125307
Zhang ZW, Guo L, Xi C, Li JH, Li Z, Peng LW, Wu MH, Ren ZY, Pan DY (2012) Highly reduced TiO2-δ nanotube arrays with enhanced visible-light absorption and room-temperature ferromagnetism. Mater Lett 69:89–91
Sun L, Li J, Wang C, Li S, Lai Y, Chen H, Lin C (2009) Ultrasound aided photochemical synthesis of Ag loaded TiO2 nanotube arrays to enhance photocatalytic activity. J Hazard Mater 171:1045–1050
Bian HD, Shu X, Zhang JF, Yuan B, Wang Y, Liu LJ, Xu GQ, Chen Z, Wu YC (2013) Uniformly dispersed and controllable ligand-free silver-nanoparticle-decorated TiO2 nanotube arrays with enhanced photoelectrochemical behaviors. Chem Asian J 8:2746–2754
Chen KS, Feng XR, Hu R, Li YB, Xie K, Li Y, Gu HS (2013) Effect of Ag nanoparticle size on the photoelectrochemical properties of Ag decorated TiO2 nanotube arrays. J Alloys Compd 554:72–79
Chen T, Wang T, Li YW, Yang YS, Jiao ZB, Ye JH, Bi YP (2014) Controllable synthesis of silver-nanoparticle-modified TiO2 nanotube arrays for enhancing photoelectrochemical performance. Nanosci Nanotech Lett 6:672–680
Valenti M, Kontoleta E, Digdaya IA, Jonsson MP, Biskos G, Schmidt-Ott A, Smith WA (2016) The role of size and dimerization of decorating plasmonic silver nanoparticles on the photoelectrochemical solar water splitting performance of BiVO4 photoanodes. Chemnanomat 2:739–747
Wang Y, Li Z, Tian YF, Zhao W, Liu XQ, Yang JB (2014) Facile method for fabricating silver-doped TiO2 nanotube arrays with enhanced photoelectrochemical property. Mater Lett 122:248–251
Macak JM, Gong BG, Hueppe M, Schmuki P (2007) Filling of TiO2 nanotubes by self-doping and electrodeposition. Adv Mater 19:3027–3031
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21621091) and Guangdong Natural Science Foundation (2016A030313845).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Xie, K., Gong, C., Wang, M. et al. Tuning Ag morphology on TiO2 nanotube arrays by pulse reverse current deposition for enhanced plasmon-driven visible-light response. J Appl Electrochem 47, 959–968 (2017). https://doi.org/10.1007/s10800-017-1093-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10800-017-1093-y