
Abstract
Aqueous rechargeable lithium-ion batteries (ARLBs) use aqueous electrolytes, which create conditions where corrosion may occur when aluminum is used as the current collector. The electrochemical stability of AA1085 in 2 M Li2SO4 and 5 M LiNO3 aqueous electrolytes was evaluated over a range of pH conditions by cyclic voltammetry, linear sweep voltammetry, and chronoamperometry. Aluminum presented high corrosion resistance at pHs 5, 7, and 9 within the stability windows of both electrolytes. At the pH 11 condition, 2 M Li2SO4 is capable of inhibiting aluminum from pitting but the inhibiting effect is not sustainable and crystallographic pitting occurs under a continuously applied anodic potential. Aluminum was well passivated against pitting in 5 M LiNO3 electrolyte at pH 11 due to the formation of a thick corrosion product barrier layer. Raman spectra depicted the presence of sulfate and nitrate anions on aluminum surface after cyclic voltammetry at pH 11. Inductively coupled plasma results showed that the amount of dissolved aluminum in electrolyte after cyclic voltammetry increases when pH increases from 5 to 11. The chemical adsorption mechanisms of sulfate and nitrate anions on aluminum were proposed to explain the dependency of electrochemical stability of aluminum on pH, anodic potential, and type of anions. The applicability of aluminum as current collector in ARLB using the 2 M Li2SO4 and 5 M LiNO3 electrolytes is discussed.
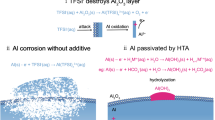
Graphical abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Lithium-ion batteries are considered as one of the most promising power sources for electric vehicles and large-scale energy storage applications. Commercial lithium-ion battery uses organic electrolytes, which provides a wide stability window of around 4 V. However, the organic solvents in the electrolyte are highly toxic and flammable, which raises significant safety concerns in the case of improper uses, such as overcharging or short-circuiting [1]. An alternative approach to the organic-based lithium-ion battery is aqueous rechargeable lithium-ion battery (ARLB), which was first introduced by Dahn’s group in the 1990s [2]. The use of aqueous electrolytes brings several benefits. It eliminates the safety issues caused by the organic electrolytes and is much more environmental friendly. Additionally, the conductivity of aqueous electrolytes is around 1 S cm−1, which is orders of magnitude higher than the typical organic electrolytes containing LiPF6. This allows higher round-trip efficiency and more flexible design of electrodes whose design thickness is often limited due to the low conductivity of the organic electrolytes. Using aqueous electrolytes could also eliminate the strict humidity controlled assembling environment required for traditional organic electrolytes thereby reducing manufacturing costs [3–5]. ARLB has not been commercialized primarily because of the narrow stability window of aqueous electrolytes, which limits energy density of the battery. The theoretical stability or operating window of an aqueous solution is 1.23 V, although the stability window was found extended to 2 V or wider due to kinetic barrier effects [6, 7]. However, the aqueous-based battery has attracted more attention recently because the advantages of ARLB make it very competitive for large-scale stationary energy storage applications where the energy density of the system is not a primary performance target [8, 9].
Aluminum is preferred as the current collector material in energy storage systems such as lithium-ion batteries and super capacitors partially because of its particular physical properties such as low density, high conductivity and low cost [10]. Degradation of the aluminum current collector may occur in organic electrolytes. For instance, serious corrosion was found on aluminum current collectors when lithium bis(trifluoromethanesulfonyl)imide [LiN(CF3SO2)2] was used as the electrolyte salt due to its acidic nature [11]. Corrosion of the current collector would significantly degrade the battery performance in the following ways: (i) it reduces the effective interfacial area between electrolyte and cathode electrode, (ii) solid corrosion products might deposit on the electrode and increase the internal impedance of battery, (iii) the dissolved species, Al3+ for instance, would contaminate electrolytes, increase self-discharge rate and impair the stability of electrodes [12–14]. Evaluating the corrosion resistance of the aluminum current collector in potential electrolytes is needed to design for battery safety and long-term performance. In the past decade, the focus of research in this area was on the corrosion behavior of aluminum current collectors under the influence of organic electrolyte chemistries, including the effects of various lithium salts, electrolyte solvent, and cathode materials [12, 14–20]. To the best of our knowledge, no evaluation has been made on the electrochemical stability of aluminum current collectors in ARLB electrolytes. Recent work has identified both 5 M LiNO3 and 2 M Li2SO4 as high performing aqueous electrolytes tested at pH 7 [21]. The reported pH values of aqueous electrolytes range from 5 to 11 so as to maintain the stability of various cathode materials, which add concerns on the risk of possible corrosion of the aluminum current collector at the higher end of the pH range [9]. Some authors speculated that the existence of sulfate and nitrate anions could inhibit the pitting corrosion of aluminum in aggressive aqueous solution by competitive adsorption [22]. An examination of the stability of aluminum in ARLB aqueous electrolytes over a range of pH values is needed to define an acceptable application window. In the present paper, the effects of pH value, applied potential and the type of anions in aqueous electrolyte on the corrosion behavior of aluminum current collector are presented. The electrochemical stability of aluminum foils in 2 M Li2SO4 and 5 M LiNO3 ARLB electrolytes is examined and the management of component corrosion during the design of energy storage systems is discussed.
2 Experimental
2.1 Electrolyte preparation
The electrolytes with different concentration and pH value were prepared in three steps. First, an aqueous solution with target pH value was prepared by dissolving specific amount of LiOH or acid into distilled water. The acid used to adjust the pH was sulfuric acid (Macron) for the Li2SO4 electrolyte and nitric acid (Acros Organics) for the LiNO3 electrolyte. The specific weight of electrolyte salts, 2 M of Li2SO4 or 5 M of LiNO3 equivalent, was added into the solution at room temperature and magnetically stirred until the salts were completely dissolved. To eliminate the effect of liquid volume expansion after the salt addition, the pH of the solution was adjusted again to the target value by gradually adding lithium hydroxide or acid until the expected pH value was achieved. The pH values of electrolytes were measured using a Mettler FE20 Ag/AgCl pH electrode.
2.2 Electrochemical measurements
Electrochemical tests were conducted using a PARSTAT-4000 in a plate material evaluating cell (BioLogic Science Instruments), which allows a constant electrode area of 0.5 cm2. Ag/AgCl with saturated KCl (0.197 V vs. SHE) solution was used as the reference electrode. The reference electrode was separated from the body of the cell using a Gamry reference electrode bridge tube with Vycor tip to prevent possible contamination from the reference electrode. The tip of the bridge tube was placed close to the working electrode to minimize the IR drop. Platinum wire, which served as the counter electrode, was shaped into a coil so a surface area approximately twice that of the working electrode was provided. Before each test, the platinum counter electrode was washed and cleaned repeatedly in dilute nitric acid followed by a rinse with distilled water. To determine the stability window of the electrolytes, linear sweep voltammetry (LSV) was performed using high-purity platinum foil as the working electrode scanned at 1 mV s−1 sweep rate from open circuit potential (OCP) either anodically or cathodically until gas evolution occurred. AA1085 foils of thickness 20 μm were evaluated in each electrolyte solution. The composition of AA1085 is presented in Table 1. AA1085 is a commercial purity aluminum alloy, which is commonly used as current collector in commercial lithium-ion batteries [23]. As-received foil samples were rinsed with isopropyl alcohol and acetone and air dried prior to testing. Cyclic voltammetry was carried out by stabilization first at OCP for 2.5 h, followed by four consecutive voltammetry cycles, starting from the negative vertex to the positive vertex of the electrolyte stability window at a 5 mV s−1 scan rate. LSV was performed by scanning from OCP to 2 V at 1 mV s−1. Chronoamperometry was taken at an anodic potential of 0.85 V for 24 h. Each measurement was performed three times using freshly cleaned aluminum samples and the representative results were reported. After the electrochemical tests, the aluminum electrode was immediately removed from the cell, gently rinsed with DI water, and dried using a gentle stream of nitrogen. All potential values are reported versus Ag/AgCl, saturated KCl scale.
2.3 Inductively coupled plasma
To further provide information on the corrosion of aluminum, the electrolyte after cyclic voltammetry was analyzed for dissolved Al3+ by inductively coupled plasma (ICP) using a Perkin Elmer optima 2100DV ICP-OES spectrometer. ICP multi-element standard solutions containing 10 and 1000 ppm aluminum were used to prepare a blank and five calibration standards of 0.01, 0.1, 1, 10, and 100 ppm aluminum. These solutions were prepared by diluting the ICP multi-element standard solution with 0.2% HNO3 in Millipore de-ionized water.
2.4 Mass spectrometer
Mass spectrometer (MS) was used to analyze the composition of the gaseous products generated by the reactions between aluminum and aqueous electrolyte after cyclic voltammetry. The MS analyses were carried out with a Hiden HPR-20 atmospheric gas analysis system. The initial conditions for the electron ionization source were set to 70 V electron-energy and 200 µA emission current. Prior to CV tests, the electrochemical cell was purged with purified argon to remove nitrogen gas for 30 min. Then the composition of the gas within the electrochemical cell was analyzed with MS as the control sample. During CV tests, the electrochemical cell was completely sealed so any gasses generated as a result of reactions between AA1085 foil and aqueous electrolyte collect within the electrochemical cell. After CV, the accumulated gas in the electrochemical cell was allowed to flow into the MS for composition analysis.
2.5 Surface characterization
The surface morphology of the electrodes after cyclic voltammetry and chronoamperometry was examined with a Hitachi S-4800 scanning electron microscopy. Raman spectroscope microscopy was performed using a Renishaw, Inc. 1000B. The Raman spectrum was excited by a helium–neon laser producing highly polarized light at 633 nm and collected in the range between 200 and 4000 cm−1. The spectra were calibrated using the 519.5 cm−1 of a piece of silicon wafer. Raman spectrum was acquired with a 10 s integration time and the power at the sample was 10 mW.
3 Results and analysis
3.1 Electrolyte stability window
LSV was used to measure the stability window of electrolytes at various pH values. The LSV curves obtained in 2 M Li2SO4 at pH 7 using platinum foil as the working electrode is shown in Fig. 1. E O and E H denote the onset potentials at which oxygen and hydrogen evolution, respectively, occurs in LSV. The measured onset gas evolution potentials and the stability window of electrolytes at pH values ranging from 5 to 11 are presented in Table 2. In Fig. 2, the measured results are compared to the equilibrium stability window of 2 M Li2SO4 and 5 M LiNO3 aqueous solutions calculated from the Nernst equation as follows:

Linear sweep voltammetry measured on Pt foil in 2 M Li2SO4 electrolyte at pH 7

Hydrogen and oxygen gassing potentials of a 2 M Li2SO4 and b 5 M LiNO3 electrolytes measured on Pt foil by LSV compared with theoretical values
All electrolytes exhibited overpotential due to the effects of kinetic barriers. The kinetic barriers are associated with making and breaking chemical bonds in the electrolysis of water, which increases the voltage required to split water at electrode/solution interface relative to the thermodynamic voltage calculated from Nernst equation [7]. At a constant electrolyte concentration, the overpotentials varied at different pH values. The span of the stability window was widest at the neutral condition and became narrower at pH conditions that deviated from the neutral value. The apparent dependence of stability window span on the pH value is consistent with the stability window results obtained by Wessells et al. using a constant current measurement method [24]. The stability windows depended primarily on the oxygen overpotential which varied with pH. The hydrogen overpotential did not contribute as significantly to the stability window though it deviated to more negative values at pHs 5 and 7 and it almost overlapped with the theoretical hydrogen evolution potentials at pHs 9 and 11 conditions.
3.2 Cyclic voltammetry
Cyclic voltammetry tests were performed to evaluate the stability of the AA1085 foil within the obtained stability windows of the aqueous electrolytes. The stabilized OCPs of aluminum measured in the aqueous electrolyte are presented in Table 3. It was noticed that in 2 M Li2SO4 electrolytes at pHs 5 and 7, and in the 5 M LiNO3 electrolyte at pH 5, the OCP was within the stability window and anodic relative to the stability window for other electrolytes. Figures 3 and 4 show the cyclic voltammetry curves measured from the first cycle to the fourth cycle. In both Li2SO4 and LiNO3 electrolytes, non-reversible oxidation peaks were present in the first cycle but the oxidation peaks diminished or receded in the following three cycles. No cathodic peak was found in the reverse scan and there were wide current plateaus in both positive and negative scans indicating that the aluminum surface remained well passivated within the scan range.

Cyclic voltammetry curves measured on Al foil in 2 M Li2SO4 electrolytes for four consecutive cycles

Cyclic voltammetry curves measured on Al foil in 5 M LiNO3 electrolytes for four consecutive cycles
In the 2 M Li2SO4 solutions of pHs 7, 9, and 11, (Fig. 3), the current density started to increase sharply in the first cycle at a specific voltage during the positive scan, followed by slight drop after a peak value was achieved and then remained at near-constant values until the positive vertex potential of the stability window. In pH 5 Li2SO4 solution, AA1085 had slightly different behavior as the current continued to increase at a slower rate after the initial peak was reached. Although the current densities at different pH values were around the same scale, it is evident that the peak positions shifted in the negative direction when the electrolyte solution became acidic or basic.
In the 5 M LiNO3 solutions (Fig. 4), aluminum exhibited a similar first cycle oxidation behavior as in 2 M Li2SO4 solutions except at pH 11. The current densities in solutions at pHs 5 and 9 were slightly larger than that at pH 7 but they remained similar. The aluminum exhibited a remarkably different rate of oxidation at pH 11. The current increased sharply beginning from the negative vertex of the scanned potential range. The current density reduced after each cycle but remained around 10−5 A cm−2, which was one order of magnitude higher compared to that at pHs 5, 7, and 9. The peak positions did not show the same pH dependency as in the 2 M Li2SO4 solutions, although the oxidation peak position at pH 11 had the most negative value.
The current plateaus in the cyclic voltammetry curves in both electrolytes reflect the concurrent dissolution and passivation phenomenon on the aluminum surface [20]. Dissolution starts when the current density increases due to the attacking of aggressive ions on aluminum, and passivation occurs right after the current density peaks. It is evident that the dissolution–passivation of aluminum follows a pH-responding mechanism in the 2 M Li2SO4 solution. When pH deviates from the neutral condition, both the dissolution and passivation processes are more easily activated, possibly due to a reduced energy barrier required for the activation process. In the 5 M LiNO3 electrolytes, there was no abrupt increase in current, and after each cycle, the current density decreased although higher current density was found in the reversible scans at pH 11. It is reasonable to assume that a fast-growing oxide or hydroxide passive film layer formed on the electrode surface during the reversible scans and the film protects the underlying aluminum matrix from further rapid dissolution.
3.3 Pitting potential measured by LSV
Many attempts have been made to obtain the critical pitting potential to evaluate the pitting susceptibility of aluminum current collectors [11, 14]. The type of anions present in aqueous electrolyte was reported to play a decisive role in determining the pitting corrosion of aluminum upon application of anodic potentials [24, 25]. LSV was employed to measure the pitting potential at which abrupt dissolution takes place. Figure 5a, b depict the linear voltammetry scanned from OCP to 2 V. The current density increased sharply when aluminum was polarized to an anodic potential in 2 M Li2SO4 solution at pH 11 and all 5 M LiNO3 solutions, with severe pitting visible on electrodes after the test.

Linear sweep voltammetry measured on Al foil in a 2 M Li2SO4 and b 5 M LiNO3 electrolytes at different pH values
In 2 M Li2SO4 electrolyte, pitting only occurred at pH 11 and the pitting potential was determined as 1.275 V. Although at pH 5 there is a current increase at 1.298 V, but it immediately declined to μA cm−2 levels and there was no sign of pitting on the electrode. In 5 M LiNO3 electrolytes, the pitting potentials for pHs 5, 7, and 9 were around 1.655 V and shifted to a more positive value, 1.734 V, at pH 11. Notably, aluminum was prone to pitting beyond the stability window in neutral 5 M LiNO3 solutions but pitting was not observed in neutral 2 M Li2SO4 solutions, which indicates a possible inhibiting effect due to the existence of sulfate anions. The inhibition of sulfate on aluminum pitting at lower pH values may be associated to the physical blocking effect at high anodic potentials [25]. The remarkable difference in pitting potential E p confirmed that the pH, anodic potential, and more importantly the type of anions in solution controlled the initiation and growth of pitting on aluminum.
3.4 Chronoamperometry
Chronoamperometry tests were carried out on aluminum electrodes at 0.85 V, a potential below the positive vertex of stability windows obtained from LSV, for a period of 24 h and are presented in Fig. 6. In 2 M Li2SO4 solution (Fig. 6a) at pHs 5, 7, and 9, the current density declined to 10−5 A cm−2 levels after the first few seconds of the test and remained stable through the remainder; current density at pHs 5 and 9 were close but slightly higher than that at pH 7. At pH 11, however, the current density gradually declined during the first 0.85 h but then increased sharply to mA cm−2 levels, almost three orders of magnitude higher than the other pH conditions. Severe pitting was visible on the electrode after the test at pH 11 and even part of the 20 μm thick foil was perforated. Figure 6b shows the data obtained in 5 M LiNO3 at the four pH values, the anodic current densities remained at a steady state of 10−6 A cm−2 level through the end of the tests. Although the current density at pH 11 was higher than other electrodes at the initial 4 h, it gradually falls even below others. The low current intensity indicated that aluminum was well passivated under the effect of concentrated nitrate anions. AA1085 presented different pitting-resistance in 2 M Li2SO4 and 5 M LiNO3 electrolytes. Based on the change of current density with time, the development of pitting in 2 M Li2SO4 electrolyte at pH 11 can be separated into three stages, which are indicated in Fig. 6a. At stage I, the hydroxide ions preferably adsorb on surface defect such as passive film flaws and intermetallic sites so metastable pits developed at a potential below the pitting potential. At stage II, stable pit growth occurs and aluminum dissolved at high rates after the incubation period at stage I. The hydrolysis of aluminum results in a reduction of pH value in aluminum pits thus the current density slowly dropped to lower values at stage III and the pitting growth is slowed down. The severe damage to aluminum foil caused by the pitting indicates that aluminum is not electrochemically stable in 2 M Li2SO4 solution of pH 11 when it is anodically polarized to 0.85 V within the stability window. However, aluminum presents good resistance to rapid localized corrosion in 5 M LiNO3 solutions at such anodic polarizing potentials at the same pH.

Chronoamperometry measured on Al foil in a 2 M Li2SO4 and b 5 M LiNO3 at different pH values for a period of 24 h
3.5 Surface morphology
3.5.1 Surface morphology after CV
The surface morphology of electrodes after CV tests in 2 M Li2SO4 electrolytes are examined and compared in Fig. 7. At low magnification (Fig. 7a), the surfaces of electrodes tested in electrolytes at pH 7 are free of any localized corrosion, although the surfaces showed slight roughness under high magnification (Fig. 7b). The surface morphology of electrodes tested at pHs 5 and 9 appeared similar to that at pH 7 thus the images thus were not presented. At pH 11, small amounts of pitting scattered on the electrode (Fig. 7c). The electrode at pH 11 exhibited more roughness compared to the other electrodes (Fig. 7d), which is ascribed to general corrosion that occurred during polarization in alkaline solution. EDS analysis (Fig. 8a, b) shows that the pits primarily formed around Al–Fe and Al–Fe–Si intermetallic particles, which are so called “circumferential” pitting. These intermetallic phase are commonly present as impurities in AA1085 alloy [19]. This indicates the occurrence of galvanic corrosion between the intermetallic and aluminum matrix in 2 M Li2SO4 aqueous solutions.

Surface morphology of Al electrodes after cyclic voltammetry in 2 M Li2SO4 at a pH 7, ×1000, b pH 7, ×50,000, c pH 11, ×1000, and d pH 11, ×50,000

EDS analysis on a Al–Fe and b Al–Fe–Si intermetallic particles in circumferential pitting formed on AA1085 after CV in 2 M Li2SO4 at pH 11
Small scattered circumferential pitting was detected on the electrode at pH 9 (Fig. 9a, b) after being reversibly scanned in 5 M LiNO3 solution. At pH 11, a uniform layer of corrosion product was found (Fig. 9c, d). At a few locations, the corrosion product layer fell off due to its expansion difference with the matrix during drying (Fig. 9e). Figure 9f shows the cross-section profile of the electrode and it clearly indicates that the thickness of the corrosion product film is around 2 μm. The formation of this corrosion product layer with considerable thickness proves that considerable amounts of aluminum was oxidized during cyclic voltammetry and the corrosion product precipitated on the aluminum surface, which correlates to the high current density obtained in cyclic voltammetry tests. Since this thick corrosion product layer was only observed in 5 M LiNO3 electrolyte at pH 11, it is deduced that the corrosion product was formed due to the coexistence of nitrate and hydroxide in that electrolyte.

Surface morphology of Al electrodes after cyclic voltammetry in 5 M LiNO3 at a pH 9, ×1000, b pH 9, ×50,000, c pH 11, ×1000, d pH 11, ×50,000, e surface corrosion products at pH 11, ×5000, and f cross-section profile of corrosion products at pH 11, ×3000
3.5.2 Surface morphology of pitting after chronoamperometry
After anodic polarization for 24 h in 2 M Li2SO4 solution with pH 11, the perforated aluminum electrode was examined under SEM and the surface morphology is presented in Fig. 10. Severe localized corrosion occurred on the electrode tested at pH 11 and part of the foil was completely corroded away (Fig. 10a). There was clear evidence of crystallographic etching with cubic morphology observed inside the pits (Fig. 10b). The presence of the geometric facets inside pits is the result of preferable attack along well-defined crystallographic directions. This form of corrosion is identical to the crystallographic corrosion with {100} facets on aluminum or aluminum alloys in citrate solution and chloride solution reported by other authors [26]. This corrosion was presumably caused by the minimum elastic modulus, lowest intensity, and lowest interatomic bonding force in this crystallographic direction of aluminum.

Morphology of pitting formed on Al electrode after chronoamperometry test in 2 M Li2SO4 electrolyte at pH 11, a ×1000 and b ×20,000
3.6 Raman spectroscopy
The different surface morphologies of AA1085 foil after electrochemical tests indicate that the composition of the surface passive films on AA1085 may be distinct in 2 M Li2SO4 and 5 M LiNO3 electrolytes at different pH values. The composition of the surface passive layer plays an important role in determining the stability of aluminum current collectors. For instance, it was reported that aluminum suffers serious localized corrosion in organic-based lithium-ion batteries containing bis(fluorosulfonyl) imide solvent [27]. However, the addition of LiPF6 in the solvent allows the formation of a low-solubility strong passivation AlF3 film on aluminum that is capable of protecting the substrate from pitting corrosion [27]. Thus, studying the structure and composition of aluminum passive film has been a research interest. Figure 11 shows the Raman spectra of aluminum electrodes after cyclic voltammetry measurements. The results are compared to the spectrum obtained from a cleaned as-received aluminum foil. The spectrum of the baseline aluminum foil presented a very broad band in the region between 600 and 1200 cm−1 with relative low intensities, which indicates that the passive film on the surface is amorphous. This is in agreement with the well-accepted knowledge that an amorphous alumina with the thickness of 2–4 nm forms on aluminum under ambient conditions and the band is assigned to amorphous alumina. For the electrodes tested in Li2SO4 electrolytes (Fig. 11a), the spectra obtained at pHs 5, 7, and 9 exhibited the same broad band between 600 and 1200 cm−1. A broad peak between 2800 and 3000 cm−1 with low intensity was detected at pHs 5 and 9, which was assigned to O–H stretching modes due to an outer layer of hydrated alumina [28, 29]. The spectrum at pH 11, however, was characterized by sharp bands at 598, 983, 1067, 1389, and 1519 cm−1. For the electrodes tested in LiNO3 electrolytes (Fig. 11b), the broad band corresponding to alumina only existed at the pH 5 condition. Bands with relatively low intensities were obtained at pHs 7 and 9. At pH 11, sharp bands presented at 718, 1058, 1392, and 1516 cm−1. A broad band also existed in the wavenumber range between 3447 and 3750 cm−1. The band positions and broadness are compared to band parameters reported in literatures and carefully analyzed [28, 30–33]. The band components, the referenced literature, and the tentative assignments for the spectrums are presented in Table 4.

Raman spectra of Al electrodes after cyclic voltammetry in a 2 M Li2SO4 and b 5 M LiNO3 at different pH values. The baseline samples shown are cleaned as-received foils not subjected to any electrochemical exposure
The analysis of Raman spectra shows that within the stability window, the amorphous alumina layer remained stable in 2 M Li2SO4 electrolytes at pH 7 and was slightly hydrated at pHs 5 and 9. Once the pH increased to pH 11, the amorphous passive film was destroyed due to the attack of OH−. Notably the band at 599 and 982 cm−1 was ascribed to the ν2 and ν1 of \({\text{SO}}_{4}^{2 - } .\) The band assignments showed the coexistence of sulfate, hydroxyl, and Al–OH bands, which indicate the adsorption of sulfate on aluminum surface. In 5 M LiNO3 electrolytes, the surface passive film remained amorphous only in acidic pH 5 condition. The bands with strong intensity at 1391 and 1515 cm−1 presented at pHs 7, 9, and 11 conditions were assigned to H2O associated to AlO4 and AlO6, which are associated to the hydrated surface and presence of hydroxide on aluminum [32]. The sharp band at 718 and 1058 cm−1 was due to the presence of nitrate on the corrosion product layer formed at pH 11.
3.7 Concentration of dissolved Al3+ after CV
Dissolved aluminum in electrolytes after cyclic voltammetry were measured by ICP and the results are presented in Table 5. In both Li2SO4 and LiNO3 solutions at pHs 5, 7, and 9, low concentrations of dissolved aluminum were detected in the electrolyte. At pH 11, 3.27 ppm of aluminum was measured in 2 M Li2SO4 electrolyte and 90.47 ppm of aluminum was measured in 5 M LiNO3 electrolyte. The amount of dissolved aluminum with the change of pH of aqueous solutions presents a trend similar to the predicted aqueous stability of crystalline and partially amorphous aluminum oxides by Tromans [34], where an increased aluminum solubility was expected when pH increases from 5 to 11. As calculated by the thermodynamic based model, the solubility of Al2O3 covered aluminum was predicted to increase at least four orders of magnitude when pH value changes from 5 to 11. The experimental results of dissolved aluminum after CV confirm that a significant higher concentration of aluminum exist in 2 M Li2SO4 and 5 M LiNO3 electrolytes at pH 11 relative to that at other pH values.
3.8 Gas composition analysis with mass spectrometer
Survey MS scans were performed on the gas collected from the Ar purged electrochemical cell before and after cyclic voltammetry tests. The mass spectrum of the gas in electrochemical cell with AA1085 as working electrode and 5 M LiNO3 electrolytes at pH 11 is presented in Fig. 12. Before cyclic voltammetry (Fig. 12a), the background mass spectrums showed a peak with high relative pressure centered at m/z = 40, which corresponds to Ar. There is a peak with low relative intensity centered at m/z = 28, which is associated to N2. The peak at mass 20 represent doubly charged argon ions, Ar2+ (m/z = 20). The presence of the small amount of nitrogen is due to the ineliminable air in the electrochemical cell after Ar purging. After cyclic voltammetry (Fig. 12b), the mass spectrum also showed the presence of Ar (m/z = 40) and nitrogen (m/z = 28). However, the relative intensity of N2 (m/z = 28) increased significantly from 14% before CV to 45% after CV. It is concluded that nitrogen gas is generated due to the reactions between aluminum foil and 5 M LiNO3 at pH 11 during cyclic voltammetry. In 2 M Li2SO4 electrolyte at pH 11, the mass spectrum of the gas in electrochemical cell is almost identical before and after the cyclic voltammetry, which indicates that no gas is generated or the amount of gas generated during CV is below the detection limit of MS.

MS spectra of gas in Ar purged electrochemical cell with AA1085 as working electrode and 5 M LiNO3 electrolyte at pH 11 a before cyclic voltammetry and b after cyclic voltammetry
4 Discussion
4.1 The inhibiting effect of anions
Anions acting as inhibitors could adsorb, compete with aggressive anions, and incorporate into passive films. This may repair passive film defects imparting better protective properties [35]. In aqueous solutions, the pitting corrosion of aluminum may be affected by sulfate and nitrate anions. Especially when the electrode is anodically polarized, sulfate and nitrate anions migrate toward the anode, adsorb on the aluminum surface, and may even penetrate the passive film [22]. It was claimed that in solutions containing chloride ions, nitrate anions could incorporate into the oxide passive film of aluminum through chemical adsorption and impede the penetration of chloride ions and mitigate pitting corrosion. The addition of sulfate anions in chloride solution may also reduce the corrosion rate of pure aluminum and change the oxide film relaxation rate [35], which is due to the competitive adsorption between sulfate and chloride anions and possible physical blocking effect of sulfate anions, although the physical adsorption was revealed only occurring on top surface.
Based on the above results and analysis, the adsorption mechanisms of concentrated sulfate and nitrate anions on aluminum are presented, specifically at slightly alkaline conditions. In the case of 2 M Li2SO4 electrolyte, severe localized corrosion only occurred at pH 11, and the existence of a considerable amount of OH− was considered to be a prerequisite to allow large-scale pitting growth on AA1085. OH− ions can attack oxide passive film and aluminum matrix by the following reactions [36, 37],
Under anodic polarization, Al metal could be oxidized into Al3+ and thus following reaction is also possible:
Due to the inevitable existence of Al–Fe and Al–Si–Fe intermetallic particles in AA1085, OH− could cause the rapid depletion of Al metal at metastable sites by the formation of local galvanic cells and detrimental pitting can evolve rapidly. As it was confirmed by Raman spectra, there was presence of sulfate on aluminum surface after cyclic voltammetry in 2 M Li2SO4 at pH 11. When sulfate ions adsorb on aluminum surface, basic aluminum sulfate preferably forms by the following reactions [38],
Sulfate anions carry a negative charge, which suggests that OH− anions would be repelled from the preferential adsorption sites, where pitting corrosion usually initiates. A competitive adsorption between \({\text{SO}}_{4}^{2 - }\) and OH− on electrode surface is expected. Under anodic polarization, the electrical energy served as the driving force to activate these reactions. Thus, a corrosion product layer comprising aluminum hydroxide–sulfate was formed. This stable, basic salt impedes the migration of OH− and prevents the further dissolution of aluminum. However, as it was found in chronoamperometry, although the current density remained at low level initially due to the basic aluminum salt, this sulfate-incorporated passive layer is not capable of inhibiting aluminum matrix over long periods under a high anodic potential. The breakdown of the basic aluminum sulfate film may be explained by its relatively low thermodynamic stability in alkaline solutions [39]. Sulfate alone would not be considered as an ideal inhibitor for aluminum due to its limited effectiveness on inhibition. However, when applied together with other inhibiting species it might play a role on raising the protective efficiency of the inhibitor package.
Nitrate affects the electrochemical stability of aluminum by a different inhibiting mechanism than with sulfate anions. The surface morphology of AA1085 after CV in 5 M LiNO3 at pH 11 showed the formation of a thick and compact corrosion product film, which could act as a barrier between aluminum metal and the electrolyte. MS results confirmed that nitrogen gas is generated due to reactions between AA1085 and 5 M LiNO3 at pH 11 during cyclic voltammetry. It was reported that in alkaline conditions, reduction of nitrate takes place with the presence of aluminum powder. The principle reaction product are aluminum hydroxide, ammonia, nitrite anion and nitrogen gas [40–42]
Ammonia was not detected in mass spectrum possibly due to its high dissolubility in aqueous solutions. Once the passive film was dissolved by the attack of OH− and fresh aluminum matrix was exposed to the electrolyte, the adsorbed \({\text{NO}}_{3}^{ - }\) and OH− will react with aluminum and aluminum hydroxide forms on aluminum surface. The vigorous formation of an aluminum hydroxide layer on the surface explains the high capacitance in the cyclic voltammetry at pH 11 in 5 M LiNO3. Growth of such a dense hydroxide film passivates aluminum surface [43], impedes the migration of aggressive anions, and results in the gradually reduced current density after each consecutive cycle. Brett et al. reported that nitrite is an effective corrosion inhibitor on aluminum in near-neutral aqueous chloride solutions [25]. The presence of \({\text{NO}}_{2}^{ - }\) due to Reaction (9) is expected to aid the inhibition effect on aluminum by the competitive adsorption between nitrite and hydroxide anions on aluminum surface. As Raman spectrum showed the presence of nitrate on aluminum in 5 M LiNO3 at pH 11, it is deduced that the chemical adsorption of nitrate occurred on the top surface of aluminum and the growth of the Al(OH)3 layer is controlled by the mass transfer across the corrosion product layer.
The concentration of dissolved aluminum ions in test solutions depends on the rate of removal of the aluminum from the metal/passive film interface toward bulk solution by diffusion. The formation of basic aluminum sulfate in 2 M Li2SO4 electrolyte and Al(OH)3 in LiNO3 electrolyte at pH 11 could explain the different amounts of dissolved aluminum after CV tests. Basic aluminum sulfate was shown to have cation-selectivity [44]. When a cation-selective diffusion layer is formed on aluminum, the ion transportation between substrate metal and electrolyte is dominated by cations. The migration of OH− through the passive film layer is impeded, which contributes to the stability of aluminum. The formation of the Al(OH)3 film in 5 M LiNO3 electrolyte is attributed to a general corrosion process over the entire aluminum surface. More importantly, this film is not ion-selective and its solubility in alkaline solution is high. Therefore, free exchange of Al3+ and OH− still occurs between the aluminum matrix and the aqueous solution until the formed layer is sufficiently thick to impede the migration of ions. These conclusions are further confirmed by comparing the results obtained in chronoamperometry tests. In 2 M Li2SO4 electrolyte of pH 11, the stability at the start of the test is primarily ascribed to the formation of the cation-selective film. In 5 M LiNO3 electrolytes, the continuous growth of the Al(OH)3 layer resulted in the gradual decline of current density.
4.2 Material selection for current collector in ARLB
According to the Pourbaix diagram, within the potential range defined by the stability window, aluminum is stable when pH value is between 4 and 8 but corrosion will occur beyond this pH range. Although it describes the fundamental thermodynamics of reactions that determines the corrosion behavior of aluminum, it does not take account of the passive film formed on aluminum that may impede aggressive anions, which may act as corrosion rate limiting step. Based on the results obtained, aluminum presented high electrochemical stability within the pH range between 5 and 9 in both electrolytes. At pH 11, the existence of high concentration of sulfate and nitrate anions both result in the formation of protective passive films, by the previously proposed inhibiting mechanisms. However, the surface layer formed in Li2SO4 could not survive under the effect of anodic potential and a rapid dissolution of aluminum current collector occurred. At the same pH condition in 5 M LiNO3 electrolytes, although the surface layer of oxide–hydroxide passive film protects aluminum well against dangerous localized corrosion, the considerable thickness of corrosion products formed by Reactions (9)–(11) could increase the resistance between the current collector and cathode active material to the detriment of cell performance. Also, a considerable amount of dissolved aluminum was detected in LiNO3 electrolyte after CV test. Considering the harmful effects of aluminum corrosion on battery performance, including (i) reduction of effective interfacial area between electrolyte and cathode electrode, (ii) solid corrosion products might deposit on the electrode and increase the internal impedance of battery, and (iii) the dissolved species contaminate electrolytes and increase self-discharge rate and impair the stability of electrodes [11–13], it risks the adverse impacts of (i) reduction of effective interfacial area between electrolyte and cathode electrode and (iii) contamination of electrolytes at pH 11 in 2 M Li2SO4 electrolyte, (ii) increased internal impedance and (iii) contamination of electrolytes at pH 11 in 5 M LiNO3 electrolyte when AA1085 is used as current collector. In these high pH conditions, the use of materials that are resistant to alkaline attack, e.g., stainless steel, may be preferred.
5 Conclusion
In aqueous electrolytes, the electrochemical stability of aluminum was influenced by the pH value, the concentrated anion, and the anodic polarization potential. The results are concluded as follows:
-
(1)
The pH value of ARLB electrolyte has a direct impact on the stability of aluminum. Under anodic polarization aluminum remains passivated in pHs 5, 7, and 9 in aqueous electrolytes. It risks severe localized dissolution at pH 11 in 2 M Li2SO4 electrolyte but it is well protected from pitting in 5 M LiNO3 electrolyte at pH 11 due to the formation of thick corrosion product barrier layer.
-
(2)
Both concentrated sulfate and nitrate anions could assist inhibiting aluminum pitting though competitive chemical adsorption with OH− in slightly alkaline solutions. Sulfate ions were incorporated into the aluminum surface passive film in alkaline solution and form an ion-selective basic aluminum salt film while nitrate ions were not.
-
(3)
Aluminum presented good electrochemical stability at pHs 5, 7, and 9 in both aqueous electrolytes. At pH 11, although sulfate and nitrate anions are capable of inhibiting aluminum pitting corrosion, however, the limited inhibiting effects of sulfate anions and the thick corrosion product layer formed in nitrate-contained electrolytes would eventually lead to deterioration of battery performance.
References
Scrosati B, Hassoun J, Sun Y-K (2011) Lithium-ion batteries. A look into the future. Energy Environ Sci 4:3287
Li W, Dahn JR, Wainwright D (1994) Rechargeable lithium batteries with aqueous-electrolytes. Science 264(5162):1115–1118
Wang Y, Yi J, Xia Y (2012) Recent progress in aqueous lithium-ion batteries. Adv Energy Mater 2:830–840
Wessells C, Huggins RA, Cui Y (2011) Recent results on aqueous electrolyte cells. J Power Sources 196:2884–2888
Song W, Ji X, Zhu Y, Zhu H, Li F, Chen J, Lu F, Yao Y, Banks CE (2014) Aqueous sodium-ion battery using a Na3V2(PO4)3 electrode. ChemElectroChem 1:871–876
Wang GJ, Zhao NH, Yang LC, Wu YP, Wu HQ, Holze R (2007) Characteristics of an aqueous rechargeable lithium battery (ARLB). Electrochim Acta 52:4911–4915
Xiang C, Papadantonakis KM, Lewis NS (2016) Principles and implementations of electrolysis systems for water splitting. Mater Horiz 3:169–173
Alias N, Mohamad AA (2015) Advances of aqueous rechargeable lithium-ion battery: a review. J Power Sources 274:237–251
Kim H, Hong J, Park K, Kim H, Kim S-W, Kang K (2014) Aqueous rechargeable Li and Na ion batteries. Chem Rev 114:11788–11827
Iwakura C, Fukumoto Y, Inoue H, Ohashi S, Kobayashi S, Tada H, Abe M (1997) Electrochemical characterization of various metal foils as a current collector of positive electrode for rechargeable lithium batteries. J Power Sources 68:301–303
Li Y, Zhang X-W, Khan SA, Fedkiw PS (2004) Attenuation of aluminum current collector corrosion in LiTFSI electrolytes using fumed silica nanoparticles. Electrochem Solid State Lett 7:A228
Braithwaite JW, Gonzales A, Nagasubramanian G, Lucero SJ, Peebles DE, Ohlhausen JA, Cieslak WR (1999) Corrosion of lithium-ion battery current collectors. J Electrochem Soc 146:448–456
Church BC, Kaminski DT, Jiang J (2014) Corrosion of aluminum electrodes in aqueous slurries for lithium-ion batteries. J Mater Sci 49:3234–3241
Zhang SS, Jow TR (2002) Aluminum corrosion in electrolyte of Li-ion battery. J Power Sources 109:458–464
Whitehead AH, Schreiber M (2005) Current collectors for positive electrodes of lithium-based batteries. J Electrochem Soc 152:A2105–A2113
Zhang X, Devine TM (2006) Identity of passive film formed on aluminum in Li-ion battery electrolytes with LiPF6. J Electrochem Soc 153:B344
Morita M, Shibata T, Yoshimoto N, Ishikawa M (2002) Anodic behavior of aluminum in organic solutions with different electrolytic salts for lithium ion batteries. Electrochim Acta 47:2787–2793
Myung S-T, Hitoshi Y, Sun Y-K (2011) Electrochemical behavior and passivation of current collectors in lithium-ion batteries. J Mater Chem 21:9891
Li SY, Church BC (2016) Effect of aqueous-based cathode slurry pH and immersion time on corrosion of aluminum current collector in lithium-ion batteries. Mater Corros 67:978–987
Yang H, Kwon K, Devine TM, Evans JW (2000) Aluminum corrosion in lithium batteries an investigation using the electrochemical quartz crystal microbalance. J Electrochem Soc 147:4399
Zhao M, Huang G, Qu F, Wang F, Song X (2015) Electrochemical performances of (LiMn0.6Fe0.4PO4/C)//LiV3O8 in different aqueous solution electrolyte. Electrochim Acta 151:50–55
Pyun S-I, Moon S-M (1999) The inhibition mechanism of pitting corrosion of pure aluminum by nitrate and sulfate ions in neutral chloride solution. J Solid State Electrochem 3:331–336
Zhao P, Chen X, Chen W, Yongzhao Z (2013) Effect of homogenization treatment conditions on the recrystallization behavior of Al–1.2Mn aluminum alloy sheets. In: Sadler B (ed) Light metals. Wiley, Hoboken, pp 341–346
Wessells C, Ruffο R, Huggins RA, Cui Y (2010) Investigations of the electrochemical stability of aqueous electrolytes for lithium battery applications. Electrochem Solid State Lett 13:A59–A61
Brett CM, Gomes IAR, Martins JPS (1994) Inhibition of aluminium corrosion in chloride media: an impedance study. J Appl Electrochem 24:1158–1163
Ma J, Wen J, Gao J, Li Q (2014) Performance of Al–1Mg–1Zn–0.1Ga–0.1Sn as anode for Al-air battery. Electrochim Acta 129:69–75
Cho E, Mun J, Chae OB, Kwon OM, Kim H-T, Ryu JH, Kim YG, Oh SM (2012) Corrosion/passivation of aluminum current collector in bis(fluorosulfonyl)imide-based ionic liquid for lithium-ion batteries. Electrochem Commun 22:1–3
Ruan HD, Frost RL, Kloprogge JT (2001) Comparison of the Raman spectra of bayerite, boehmite, diaspore and gibbsite. J Raman Spectrosc 32:745–750
Bunker BC, Nelson GC, Zavadil KR et al (2002) Hydration of passive oxide films on aluminum. J Phys Chem B 106:4705–4713
Kloprogge J, Frost R (1999) Raman microscopy study of basic aluminum sulfate. J Mater Sci 4:4199–4202
Ahern AM, Schwartz PR, Shaffer LA (1992) Characterization of conversion-coated aluminum using Fourier transform infrared and Raman spectroscopies. Appl Spectrosc 46:1412–1419
Cherepy NJ, Shen TH, Esposito AP, Tillotson TM (2005) Characterization of an effective cleaning procedure for aluminum alloys: surface enhanced Raman spectroscopy and zeta potential analysis. J Colloid Interface Sci 282:80–86
Kloprogge JT, Frost RL (1999) Raman and infrared microscopy study of zunyite, a natural Al13 silicate. Spectrochim Acta A 55:1505–1513
Tromans D (2005) Thermodynamic evaluation of the effects of amorphism on film breakdown and pitting initiation. J Electrochem Soc 152:B460
Amin MA, Abd El Rehim SS, El-Lithy AS (2010) Pitting and pitting control of Al in gluconic acid solutions—polarization, chronoamperometry and morphological studies. Corros Sci 52:3099–3108
Doche ML, Rameau JJ, Durand R, Novel-Cattin F (1999) Electrochemical behaviour of aluminium in concentrated NaOH solutions. Corros Sci 41:805–826
Adhikari S, Hebert KR (2008) Factors controlling the time evolution of the corrosion potential of aluminum in alkaline solutions. Corros Sci 50:1414–1421
Foley RT, Nguyen TH (1982) The chemical nature of aluminum corrosion. J Electrochem Soc 129:464–467
Povar I, Spinu O (2014) The role of hydroxy aluminium sulfate minerals in controlling Al3+ concentration and speciation in acidic soils. Cent Eur J Chem 12:877–885
Murphy AP (1991) Chemical removal of nitrate from water. Nature 350:223–225
Fanning JC (2000) The chemical reduction of nitrate in aqueous solution. Coord Chem Rev 199:159–179. doi:10.1016/S0010-8545(99)00143-5
Foley RT, Nguyen TH (1982) The chemical nature of aluminum corrosion: II. The initial dissolution step. J Electrochem Soc 129:27–32
Wang J-B, Wang J-M, Shao H-B et al (2007) The corrosion and electrochemical behaviour of pure aluminium in alkaline methanol solutions. J Appl Electrochem 37:753–758
Dan Z, Muto I, Hara N (2010) Role of corrosion products in the suppression of atmospheric corrosion of aluminum and its alloys. ECS Trans 25:23–33
Kloprogge J, Frost R (1998) Raman microscopy study of basic aluminium nitrate. Spectrochim Acta A 55:163–169
Acknowledgements
Electron microscopy was carried out at UW-Milwaukee Biological Sciences Microscopy Center Raman Spectroscopy was conducted at the UW-Milwaukee Advanced Analysis Facility.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Li, S., Church, B.C. Electrochemical stability of aluminum current collector in aqueous rechargeable lithium-ion battery electrolytes. J Appl Electrochem 47, 839–853 (2017). https://doi.org/10.1007/s10800-017-1081-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10800-017-1081-2