
Abstract
Iron(III) oxide tablets were electrolytically reduced to iron in molten sodium hydroxide at 530 °C and recovered to produce iron with 2 wt.% oxygen suitable for re-melting. The cell was operated at 1.7 V and an inert nickel anode was used. The thermodynamics and mechanism of the process was also investigated. By controlling the activity of sodium oxide in the melt, the cell could be operated below the decomposition voltage of the electrolyte with the net sequence of events being the ionization of oxygen, its subsequent transport to the anode and discharge leaving behind iron at the cathode. A reduction time of 1 h was achieved for a 1 g oxide tablet (close to the theoretical reduction time predicted by Faraday’s laws) at a current density of 520 mA cm−2 with iron phase yields of ∼90 wt.%. The energy consumption was 2.8 kWh kg−1.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Directives to reduce global emissions in CO2 [1] are now firmly in place which puts pressure on heavy industry sectors such as iron and steel whose products are used in vast quantities around the world.
There is a European directive to partially replace existing technology with “greener” routes. A process is proposed in this paper based on the electro-deoxidation of a metal oxide in a molten salt electrolyte which has oxide ion solubility. The pioneering work in this field, the so-called FFC process [2] describes the electro-deoxidation of titanium dioxide in a calcium chloride electrolyte at 950 °C. The process involves the direct electrochemical reduction of a metal oxide in molten calcium chloride. The oxide cathode is in good physical contact with a current collector and the metal oxide is progressively reduced through a series of part reactions involving intermediate binary oxide phases of the form aCaO:bMxOy where a, b, x and y are integers and M is a metal. Oxide ions are released into the bulk salt and discharged on a graphite anode which is consumed to form CO2.
The work in this paper applies the FFC process to the reduction of iron(III) oxide (Fe2O3) in a sodium hydroxide electrolyte at the lower temperature of 530 °C to produce a grade of iron (<10 wt.% oxygen) that requires minimal re-smelting thus forming much less CO2. It is possible to power such a process from renewable energy sources.
The electrolyte can be used to perform many batch reductions before replacement is necessary and the only by-product is oxygen which is formed on the anode. This makes the process chemistry much more cleaner and controllable than with calcium chloride and graphite anodes where halocarbon compounds are formed along with CO2 and carbon-fouling of the salt which leads to poor control and reproducibility. Furthermore, co-deposition of calcium and carbonates in calcium chloride severely reduces the current and energy efficiency of the process.
There appears to be no viable aqueous or organic electrolyte alternative to the molten salt electrolysis route because the space-time yield and current densities in such cells are too small for industrial application.
Haarberg et al. [3] have studied electro-deoxidation of iron oxide in calcium chloride. In the best of their experiments, they produced a phase of iron that was 5 mol% in oxygen using a nickel ferrite/copper cermet anode. However, the latter decomposed severely when using viable current densities, resulting in anodic dissolution of copper into the salt and subsequent co-deposition and contamination of the iron phase. When graphite anodes were used, the reduction was hampered by carbon particles blocking the pores of the oxide preventing electrolyte contact to the oxide.
2 Experimental
2.1 Precursor preparation
Iron(III) oxide (Aldrich 99+%) of particle size between 1 and 5 μm was pressed without a binder at 3 ton in an isostatic press using a 20 mm dye. The un-sintered tablets were weighed and their volume measured. The tablets were then sintered at various temperatures in the range 900–1300 °C for 30 min with a ramp rate of 3 °C/min was used. After sintering, the tablets were re-weighed and their volume re-determined. This enabled the pre-sintered and sintered densities to be estimated based on a uniform pore distribution.
2.2 Materials and setup
A nickel (201 alloy—99% Ni with Cu and Fe added to prevent embrittlement at temperatures above 315 °C) basket with a screw-in nickel connector rod was used to hold the tablet throughout an experiment. The crucible (55 mm id × 61 mm od × 160 mm height) was also fabricated from nickel and contained in an outer alumina crucible. The anode consisted of a nickel 201 rod of 5 mm diameter.
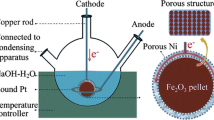
The crucible was packed with ∼400 g of sodium hydroxide (97% assay) pellets (∼6 × 4 mm). The salt was melted and “topped-up” to 600 g. The purpose of the large pellets was to reduce the surface area, reducing water absorption during transport to the crucible and furnace. The entire assembly was held in a sealed reactor made from Inconel 600™ which in turn sat inside a vertical tube furnace. The reactor shown in Fig. 1 was filled with argon and a steady flow (50 cm3 min−1) maintained throughout the experiment.

Schematic of high temperature cell
2.3 Method
Reductions using an iron(III) oxide cathode and nickel anode were performed. A power supply (Thurlby Thandor PL154) with feedback sensing was used which enabled the current to be accurately measured across a counter resistor and maintain a constant applied voltage. A digital data recorder (Agilent 34970A) was used to record voltage, current and temperature.
The anode was submerged 5 cm into the salt and the cathode tablet 7 cm. The system was allowed to reach thermal equilibrium by waiting 15 min prior to applying a voltage which was slowly swept to the target value to prevent spiking of the current.
After the reduction, the Ni basket was raised near the top of the reactor (gas temperature ∼90 °C) and left to cool in argon. The powder/tablet and basket were then removed and immersed in cold de-aerated water to dissolve the electrolyte and recover the product. In later experiments, water was replaced with aqueous ethanol (90 vol.% ethanol:10 vol.% water) to help prevent partial re-oxidation of the iron. After several washes, the pH checked using phenolphthalein pH indicator paper to detect the presence of alkali, the rod was removed from the basket and the tablet or powder dried in argon at room temperature. The effect of cell voltage and adding 1 wt.% sodium oxide to the electrolyte were investigated.
The decomposition voltage of the electrolyte containing 1 wt.% Na2O was measured by applying a staircase voltage to a cell containing an empty nickel basket and observing the current. Tangents were drawn to the curves representing current flow before and after salt decomposition and the point of intersection extrapolated to the voltage axis to determine the dynamic decomposition voltage of the electrolyte.
The products were characterised by Scanning Electron Microscopy (SEM, JEOL 5800), Electron Dispersion Analysis by X-Rays (EDAX Oxford Instruments INCA x-sight 6587) and X-Ray diffraction analysis (XRD Phillips X’Pert PW3020) and Inductively Coupled Plasma Spectrophotometry Atomic Emission Spectrophotometry) ICPAES (Varian axial Liberty Series II).
3 Results and discussion
3.1 Sintering
Figure 2 shows an approximate inverse linear trend was observed between sintering temperature and post-sintered porosity. Tablets sintered at 1050 °C or less tended to form powder when reduced, whereas those sintered at temperatures greater than 1050 °C were of lower porosity but tended to remain intact after the reduction. A maximum shrinkage of 43 vol.% was observed when the sintering temperature was 1300 °C, and just 8.5 vol.% when sintered at 900 °C.

Effect of sintering temperature on porosity of iron(III) oxide tablet
3.2 Core parameter investigation
Table 1 summarises the experiments performed, visual observations of the product, iron phase purity and other phases present as determined by X-ray diffraction analysis. In the latter, a semi-quantitative analysis was performed by determining the scale factor for each of the analytical peaks and comparing relative intensity ratios (RIRs) from standard reference material spectra. A normalized RIR method was used to determine the phase compositions.
3.3 Electrodeoxidation and salt decomposition
Table 1 shows that NaFeO2 spontaneously forms when the Fe2O3 tablet is added to molten NaOH and so becomes the starting material for electro-deoxidation:
The decomposition voltage at unit activity (Eo) of NaFeO2 is 1.368 V assuming unit activity of Na2O in the salt at 530 °C:
The equilibrium constant Keq of the dissociation of NaOH dictates the initial activities of Na2O and H2O:
At 530 °C, K eq = 2.645 × 10−11 and:
The term \( (a_{{{\text{NaOH}}}} )^{2} \) is unity so that initially, \( a_{{{\text{H}}_{{\text{2}}} {\text{O}}}}=a_{{\text{Na}}_{{\text{2}}} {\text{O }}}=\sqrt {K_{{\text{eq}}} }={\text{5}}{\text{.143}} \times {\text{10}}^{{ - {\text{6}}}} \) and based on the Lux-Flood scale [4] the salt is neutral. Allowing for the presence of dissolved Na2O using the Gibbs function with pure Na2O (the solute) as the standard state:
The addition of this term to the ΔG° values enables the ΔG for any reaction to be calculated and also the decomposition potential from:
where z is charge transferred, E is the potential and F is Faraday’s constant. When the oxide tablet is added to the salt, the local activity of Na2O will be low due to reactions (1) and (2) and the salt becomes a strong Lux-Flood acid. So E° for NaFeO2 is decreased and gradually increases during electro-deoxidation as Na2O is regenerated as shown in reaction (3). The activities of Na2O and H2O in the salt will also dictate their respective decomposition voltages and can be calculated in a similar way. There are two electrolyte decomposition routes both sharing the same reaction at the anode;
The corresponding part reactions at the cathode are: Case 1 where water is decomposed:
Case 2 where sodium hydroxide is decomposed (as in the Castner process [5]):
Case 3 where NaFeO2 is reduced by electro-deoxidation:
Table 2 summarises the calculated changes in salt composition at 530 °C during electro-deoxidation and the respective component decomposition voltages at various H2O, Na2O and NaFeO2 activities. The activity of Na2O under acidic conditions approaches zero which can be inferred from the number of moles Fe2O3 in the tablet and Eqs. 1 and 2. A Na2O activity of 10−11 was used based on the work of Trémillon [6] who states that in strongly acidic solutions [H2O] and aH2O approach 1 where the ionic product is 10−11.
Under strongly basic conditions the activity of the 1 wt.% Na2O in the salt was estimated from the phase diagram in Fig. 3. \( a_{{{\text{Na}}_{{\text{2}}} {\text{O}}({\text{s}})}} = 1 \) when the salt contains 27 wt.% sodium oxide or mole fraction of 0.193 at the saturation point X on the diagram. For a 1 wt.% addition of sodium oxide, the mole fraction (X1 wt.%) of the solid is 0.00647. The free energy of freezing is given by:

Phase diagram for the NaOH:Na2O system [9]
Since the slope of the liquidus approximates a straight line over the 1 wt.% to 27 wt.% composition, the activity coefficient is assumed constant over this range. Taking the free energy of freezing ΔG to be −21.09 kJ mol−1 at 530 °C, \( a_{{{\text{Na}}_{{\text{2}}} {\text{O}}(\text{l})}} = 23.5 \) when \( a_{{{\text{Na}}_{{\text{2}}} {\text{O}}(\text{s})}} = 1 \) for a 27 wt.% addition, so for a 1 wt.% addition:
Figure 4 shows the current–voltage plot to determine the decomposition voltage of the electrolyte under strongly basic conditions. The dynamic decomposition voltage (i.e. the minimum voltage necessary to perform an electrolysis) may be extrapolated from the intersection of line AB and CD and yields a value of 1.80 V. This value will include the charge transfer overvoltage and when the iR drop is subtracted yields a value of 1.55 V.

IV curve for determination of decomposition voltage of strongly basic electrolyte
Table 2 shows that hydrogen evolution will occur concurrently with electro-de-oxidation under acidic and near-neutral conditions. When Na2O is added, the H2O decomposition voltage is increased to 1.64 V, beyond the applied cell voltage of 1.70 V (1.45 V with iR drop subtracted) raising the current efficiency. Sodium deposition (1.36 V) might be expected to occur as well and yet the current efficiency (∼89%) and Fig. 4 suggests this does not occur which may be due to significant charge transfer overpotentials on the ferrite cathode and the nickel anode.
The current efficiencies are calculated and shown in Table 1. It can be seen that with the addition of Na2O, high current and energy efficiencies are obtained. Furthermore, since neither hydrogen nor sodium will reduce NaFeO2 at 530 °C the mechanism of reduction must be ionisation of oxygen and its transport out of the oxide into the salt and subsequent discharge of O2 on the anode.
The current efficiency value of 89% is also supported by Fig. 4 where the background current was ∼0.1 A at an applied voltage of 1.7 V (without oxide tablet) and the cell current at this voltage during oxide reduction was typically 1.1 A which implies a current efficiency of ∼90%.
At an applied voltage of 1.40 V (1.325 V with iR drop subtracted) there is insufficient driving force to fully reduce NaFeO2. At 1.70 and at 2.00 V without the Na2O addition, hydrogen evolution parasitizes electro-deoxidation. At 1.6 V with Na2O addition, hydrogen discharge becomes less dominant which raises the current efficiency, but as the reversible voltage of NaFeO2 is approached the overvoltage becomes important, the cell current diminishes resulting in longer reduction times.
3.4 Mechanistic considerations
Table 1 shows several intermediate phases based on NaxFeyOz which suggests NaFeO2 is not reduced in one step but incorporates Na2O, Na2O2 and NaO2 into its structure. The latter phases were also observed in the XRD analyses of partially reduced sodium ferrite some of which are presented in Table 1 implying that the mechanism of reduction is more complex than that observed with electro-deoxidation of iron oxide in CaCl2 where CaO is the only dissolved oxide. The transport of oxygen out of the oxide lattice into the salt only occurs after the formation of sodium ferrites and sodium oxide. This will happen through a series of solid-state reconstructive transformations of sodium ferrite species. Similarly, calcium metallites and metallates have been reported using CaCl2 [7, 8]. Note that:
All of the above ferrite and oxide phases were detected by XRD. This leads to two possible mechanisms of reduction: Case 1 involving just Na2O:
Case 2 involving Na2O and NaO2;
The oxygen rich elimination product NaO2 was repeatedly identified by XRD in the reduction products and may form on the anode by oxidation of O2−:
The superoxide ion can migrate to the cathode and react with sodium ferrite:
and is then eliminated from the ferrite and reduced back to the oxide ion on the cathode:
It is not possible to assign decomposition voltages in case 2 due to lack of thermodynamic data for this set of sodium ferrites. The high current efficiency would imply that the mechanism shown in case 1 predominates.
Tremillion [6] has shown that superoxides form particularly in Lux-Flood basic solutions of NaOH at sufficiently high potentials on the anode. In terms of mass balance, there is no net loss or gain from the electrolyte, just discharge of oxygen removed from the iron oxide.
4 Conclusion
Iron(III) oxide (Fe2O3) was successfully reduced to iron in molten sodium hydroxide at 530 °C by the method of electro-deoxidation. The reduction can be performed below the decomposition voltage of the electrolyte without discharge of hydrogen or sodium by a prerequisite controlled addition of sodium oxide (Na2O). The optimal operating cell voltage is 1.7 V. The current efficiency of the cell under these conditions was approximately 90%.
An inert nickel anode was successfully used throughout all of the experiments, a thin but highly protective and electronically conducting nickel oxide film forming on the surface.
The starting material for electro-deoxidation is NaFeO2 formed by spontaneous reaction of Fe2O3 with the electrolyte. Below the decomposition voltage of the salt, when electro-deoxidation is predominant, there is no net change in electrolyte composition, the oxygen from the Fe2O3 being mass balanced by discharge on the anode by oxidation of hydroxyl ions to form oxygen and water.
The mechanism proceeds by solid-state transformations of the sodium ferrite lattice and stepwise reduction of Fe3+ to Fe2+ to Fe with elimination of sodium oxide species based on the general formula NaxOy into the electrolyte. The terminal oxides are FeO and/or [2Na2O:FeO] and [Na2O2:FeO] which are reduced directly to iron. The overall reactions on the electrodes are:
Controlling the sodium oxide activity in the cell allows the operating voltage window to be varied which may allow oxides of metals less stable than H2O and Na2O (at their residing activities) to be produced.
Scale-up of the process is very promising. The usual problems associated with high temperature electrolysis such as materials degradation and toxic gas emissions are virtually eliminated with this cell. The power consumption of the cell is 2.8 kWh kg−1 based on a cell operated at 1.7 V for 1 h. The current density was about 0.5 A cm−2 indicating that the rate of reaction was far higher than that obtained in aqueous solutions. There is no CO2 output from the cell compared to 1.18 ton CO2/ton iron for the iron blast furnace based on the reaction:
and the whole process could be powered by renewable sources.
References
Industries (2006) E.C.o.I.a.S
Chen GZ, Fray DJ, Farthing TW (2000) Nature 407:361–364
Burheim OS (2005) Institutt for Material Teknologi, University of Trondheim
Plambeck JA (1976) Encyclopedia of electrochemistry of the elements. Marcel Dekker
Newell LC (1904) Descriptive chemistry. Heath
Tremillon B (1971) Pure Appl Chem 25:395
Schwandt C, Fray DJ (2005) Electrochimica Acta 51:66–76
Yan XY, Fray DJ (2005) J Electrochem Soc 152:D12–D21
Bouaziz R, Papin G, Rollet AP (1966) C R Acad Sci Ser C 262:1053
Acknowledgements
The authors are grateful to the EPSRC for funding (Platform Grant GR/S58447/01) the work in this project.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Cox, A., Fray, D.J. Mechanistic investigation into the electrolytic formation of iron from iron(III) oxide in molten sodium hydroxide. J Appl Electrochem 38, 1401–1407 (2008). https://doi.org/10.1007/s10800-008-9579-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10800-008-9579-2