
Abstract
This work describes the performance of a tyrosinase/nickel oxide nanoparticles/glassy carbon (Tyr/NiO NPs/GC) electrode. This electrode was prepared by first applying a NiO NPs electrochemical deposition onto the GC electrode surface and then tyrosinase immobilization was applied to the surface of electrodeposited NiO NPs. Scanning electron microscopy (SEM) and atomic force microscopy (AFM) procedures demonstrated the existence of different NiO NP geometrical structures. These geometrical structures could lead to better immobilization of proteins on their surfaces. The copper containing enzyme tyrosinase successfully achieved electrical contact with the electrode because of the unique structural alignment of tyrosinase enzyme on the nanometer-scale nickel oxide surfaces. This method could be suitable for application to nanofabricated devices facilitating better performance. It was concluded that tyrosinase can be effectively applied to nanometer-scale nickel oxide surfaces.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
There are many points of intersection between nanoscience, nanotechnology and the biological sciences. The organization of metal or semiconductor nanoparticles (NPs) on surfaces has attracted substantial research efforts, many of which have been directed toward developing optical or electronic sensor systems and devices [1]. Because of their high surface-to-volume ratio and tunable electron transport properties, which are attributed to the quantum confinement effect, nanostructures can be used to facilitate both efficient electron transport and optical excitation. These two factors make them critical to the effective functioning and integration of nanoscale devices [2].
Bioelectrochemistry is an interdisciplinary field, which combines biotechnology with the electrochemical discipline. However, biomolecules display nanoscale dimensions that are comparable with the dimensions of metal or semiconductor nanoparticles (NPs). This size resemblance enables their unique photonic, catalytic and electronic properties to be merged. Extensive research efforts have been made over the past few years to design biomolecule-NP hybrid assemblies and incorporate them into the construction of biosensors, nanoscale circuitry or nanodevices [3, 4].
Enzymes are highly specific catalytic units that have been used in a variety of ways in nanotechnology. Some examples include the use of enzymes as logic agents [5], the enzyme-induced generation of NPs [6], the “cutting and pasting” of DNA [7] and other applications [8]. One significant enzyme is tyrosinase, which converts tyrosine to l-DOPA. Tyrosinase is the principal enzyme for biosynthesis of the pigment melanin and exhibits unusual kinetic behavior. It is considered to be an important biocatalyst in the neural response system as well as in the progress of some diseases, such as Parkinson’s disease [9]. Elevated tyrosinase levels have especially been observed in certain kinds of melanoma cells and its possible value as a marker for these melanoma cells has been discussed [10]. The first publication on direct electron transfer (DET) between tyrosinase and graphite appeared in 1996 [11]. Yarpolov et al. described the electrochemistry of native, holo- and apoenzyme-modified graphite electrodes using cyclic voltammetry. Various methods have been described for tyrosinase immobilization on a range of suitable substrates using conventional electrode materials as substrates. These include silver [12], glassy carbon [13, 14], graphite-epoxy resin [15], gold [16], boron-doped diamond [17], core-shell magnetic nanoparticles supported on a carbon paste electrode [18], ZnO nanoparticles [19] in addition to other materials [20–24]. Some reports also relate to the electrochemical determination of kinetic parameters of mushroom tyrosinase [25].
Several studies have been devoted to examining the potential of nickel electrochemistry for a number of applications. It has been utilized as the basis for models studying oxygen-evolution reactions [26] and the oxidation of organic compounds on passive nickel anodes [27]. It has also provided the electroanalytical approach on which sulphide interaction with an electrochemically generated nickel oxide layer is based [28, 29]. Furthermore, nickel electrochemistry has been employed in anodic stripping voltammetry for sub-speciation of Ni3S2, NiS and NiS2 in a mixture of carbon paste electrodes in acetate buffer [30] as well as for sulphide detection on a carbon substrate using electrochemically deposited nickel microparticles [31]. Other reported applications include the modification of glassy carbon electrodes by nickel oxide nanoparticles for the immobilization of hemoglobin, catalase and cytochrome c [32–35], together with the use of Ni/NiO in preparing a silicon nanotube array/gold electrode for the direct electrochemistry of cytochrome c [36].
This paper describes an electrochemical investigation of the redox reaction of immobilized mushroom tyrosinase on the Tyr/NiO NPs/GC electrode. This is the first experimental study concerning tyrosinase immobilization on NiO NPs surfaces.
2 Experimental sections
2.1 Chemicals and reagents
Tyrosinase (T 7755, from mushroom) [9002-10-2] was purchased from Sigma. The phosphate buffer solution (PBS) consisted of a potassium phosphate solution (KH2PO4 and K2HPO4 from Merck; 0.05 M total phosphate) at pH 7.0. An acetate buffer solution (CH3COONa and CH3COOH from Merck; 0.05 M) was freshly prepared. Ni (NO3)2·6H2O and the other reagents were reagent grade materials from Merck. Epinephrine was obtained from Daru Pakhsh Co. (Tehran, Iran). Deionized water was used to prepare all solutions and to rinse the electrodes.
2.2 Instruments
All electrochemical experiments were performed using an Autolab Potentiostat PGSTAT 30 (Eco Chemie, Netherlands), equipped with GPES 4.9 software. The Q value for each point of the continuous CVs was obtained from the equation; Q = \( {\int_0^t {i{\text{dt}}} } \), Q 1 = i 1Δt, Q 2 = (i 1 + i 2) Δt, Q n \( = {\sum\limits_{k = 1}^{k = n} {} } \) i k Δt. Then, the plots of E vs. Q were created.
A three-electrode cell was also used, employing a glassy carbon (GC, 2 mm in diameter) electrode or modified-GC electrodes, acting as the working electrodes. A macroscopic electrode was required to attain a sufficiently large tyrosinase sample to yield detectable direct oxidation and reduction currents. A platinum wire was applied as the counter electrode and an Ag/AgCl/KCl (sat.) was used as the reference electrode. All potentials were reported with respect to this reference. The rest of electrochemical equipment has been described earlier [37–39]. All experiments were performed at 25 ± 1 °C.
Scanning electron microscopy images were recorded using a ZEISS DSM 960, while atomic force microscopic studies were performed with the help of a DME (controller, Dual Scope C-21) and scanner (DS 95-50).
2.3 Passivation of nickel oxide nanoparticles and tyrosinase immobilization
Prior to tyrosinase immobilization, surface of the GC electrode was polished by alumina powder (1.0, 0.3 and 0.05 μm in diameter, respectively) on a polishing cloth, followed by a thorough deionized water rinsing. The electrode was then successively sonicated in ethanol and doubly distilled water to remove the adsorbed particles. Cyclic scans were carried out in PBS (0.05 M, pH 7.0) in the potential range from −0.50 to 1.0 V, until repetitive cyclic voltammograms (CVs) were obtained. The solution, in which the nickel deposition was conducted, typically consisted of a 15 mL acetate buffer (pH 4.0). The nickel was initially electrodeposited (−0.60 V, 4 min deposition time) on a GC electrode from a 1 mM nickel nitrate pH 4.0 acetate buffer solution. Afterwards, the Ni-GC electrode was placed into a fresh PBS (pH 7.0) and was electrochemically passivated with the potential cycling method (protocol for the NiO NPs/GC electrode) [28, 29, 31–35].
It has been previously demonstrated that Ni(OH)2 chemical dissolution and, hence, the amount of formed passive nickel oxide depends on solution pH. In acidic solutions, the passive film is a mixed nickel oxide (NiO(Ni2O3)) [29]. The nickel oxides (NiO and Ni3O4) are stable species [28]. Stable voltammograms were observed after continuous CVs. By changing the buffer solution, at a pH value of 4.0, from acetate to phosphate, no significant effects were observed on passivation, thereby showing that the passivation was anion independent [29].
In another protocol, the Ni-GC electrode was placed into fresh PBS including 5 mg mL−1 tyrosinase. The potential was then kept at −0.60 V for 20 min (protocol for the Tyr/NiO NPs/GC electrode). For excess tyrosinase immobilization and passivation, the potential cycling method was then applied in this condition (20 scans) from 0.75 to −0.5 V with the potential scan value of 100 mV s−1. Finally, the modified electrode was washed in deionized water and placed in PBS (pH 7.0) in a refrigerator (4–6 °C), before being employed in electrochemical measurements as the working electrode.
3 Results and discussions
3.1 Scanning electron microscopic and atomic force microscopic studies of NiO NPs
As is well known, the properties of a broad range of materials and performance of different devices depend strongly on their surface characteristics. For instance, the surface of a biomaterial/biomedical device meets the physiological environment, immediately after it is placed in the body or bloodstream. The initial contact regulates its subsequent performance [40].
Figure 1a shows the SEM image of GC electrode surface before construction of the nickel oxide nanoparticles. Figure 1b illustrates the SEM image of nanometer-scale nickel oxide particles, generated on the GC electrode surface in various sizes. Because the surface-to-volume ratio increases with size decrease, the smaller nanoparticles are able to play a very important role during the immobilization process.

(a) SEM image of the GC electrode surface before construction of the nickel oxide nanoparticles, (b) SEM image of electrodeposited nickel oxide nanoparticles on the GC electrode surface
In practice, the SEM technique has some limitations for experiments at higher resolutions. Also, SEM results only suggest the existence of nanoparticle structures and cannot map the structure of these nanoparticles in detail. To overcome these deficiencies, another nanotechnological technique was employed. In order to investigate the structure of these nanoparticles, especially the smaller ones which the SEM studies could not assess, atomic force microscopy (AFM) was used and proved to be an excellent alternative.
Figure 2 shows the AFM images of nanometer-scale nickel oxide particles. An extensive sample examination with the help of SEM and AFM techniques revealed the existence of different geometrical oxide particle shapes. Figure 2c shows an image profile for the selected direction corresponding to the arrow from Fig. 2a. These profiles reveal that diameter of the nanoparticles progressively narrows from bottom to top (like the shape of a needle). This geometrical structure demonstrates significant utility in enhancing the immobilization of proteins on its surface. According to our results from the SEM and AFM procedures, the diameter of nanometer-scale nickel oxide particles was about 80–850 nm (however, some of them had a diameter of about 1.0 μm).

(a, b) The AFM images of the electrodeposited nickel oxide nanoparticles on the GC electrode surface, (c) obtained image profile for selected direction by arrow from Fig. 2a
3.2 Direct voltammetric behavior of the Tyr/NiO NPs/GC electrode
Figure 3a shows the CVs of a Tyr/NiO NPs/GC electrode in a fresh PBS, containing 5 mg mL−1 tyrosinase. The anodic and cathodic peak currents remain unchanged with increased cycling, indicating that tyrosinase completely covers the NiO NP surfaces during the previous step (protocol for the Tyr/NiO NPs/GC electrode). This phenomenon became more evident from the plot of E vs. Q for CVs (Fig. 3b). In this figure, the distances among the rings remain more or less stable with the charge (Q) increase. For this reason, no excess immobilization took place during the potential cycling. Therefore, there was no increase in the anodic and cathodic peak currents during continuous CVs. The redox peaks in this figure are related to electrochemical oxidation and reduction of the immobilized tyrosinase on the NiO NP surfaces. In agreement with the voltammograms in Fig. 3a, it is concluded that the nanometer-scale nickel oxide particles may play a key role in the tyrosinase CV response. These nanoparticles illustrated significant influence on the electron exchange between tyrosinase and the GC electrode. Moreover, Fig. 3c demonstrates that there are no voltammetric responses on both the bare GC and NiO NPs/GC electrodes. This shows that the GC and NiO NPs/GC electrodes are electroinactive.

(a) The continuous CVs of the Tyr/NiO NPs/GC electrode into a fresh PBS (pH 7.0, 0.05 M) including 5 mg mL−1 tyrosinase, for the excess immobilization of tyrosinase and passivation, scan rate; 100 mV s−1, (b) the potentials (E) versus the charge passed through the electrochemical cell (Q) during the CVs of Fig. 3a, (c) CVs in PBS (pH 7.0, 0.05 M) at the GC electrode and the NiO NPs/GC electrode, scan rate; 50 mV s−1
The ability of the immobilized tyrosinase to exchange electrons with the NiO NP surfaces was assessed by voltammetry in a fresh PBS (pH 7.0, 0.05 M).
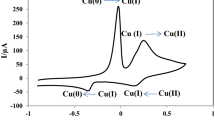
In the voltammograms (Fig. 4), tyrosinase on the Tyr/NiO NPs/GC electrode shows a reductive peak (C1) at −275 mV and a corresponding oxidative peak (A1) at 67 mV (at 10 mV s−1). The difference between the anodic and cathodic peak values was ΔE = 342 mV. These redox peaks are attributable to the redox reaction of tyrosinase electroactive centers. The formal potential (E°) for the tyrosinase redox reaction on the Tyr/NiO NPs/GC electrode was −104 mV with respect to the reference electrode (95 mV versus NHE). The formal potential value for tyrosinase on the graphite electrode was 550 mV versus SCE (794 mV versus NHE) [11]. The formal potential of tyrosinase, determined through the redox titration, was about 600 mV [41]. This value was 415 mV versus NHE for the adsorbed tyrosinase on silver [12].

The CVs of Tyr/NiO NPs/GC electrode in PBS (pH 7.0, 0.05 M) at various scan rates, from inner to outer; (a) 10, 20, 30, 40 and 50 mV s−1, the relationship between the peak currents (i pa, i pc) versus (b) the scan rates and (c) the square root of scan rates
Chemical and spectroscopic studies of tyrosinase have indicated that its binuclear copper active site can be prepared in several forms: met-tyrosinase, oxy-tyrosinase and deoxy-tyrosinase. Most of the enzymes in a freshly prepared sample (resting tyrosinase) are in the met-tyrosinase form, unable to bind O2. Only a small fraction is present in the sample as oxy-tyrosinase [42].
To further investigate tyrosinase characteristics on the Tyr/NiO NPs/GC electrode, the scan rate effect on the tyrosinase voltammetric behavior was carefully studied. Figure 4b and c show that the redox peak currents increase linearly with scan rate between 200 and 3,500 mV s−1. The correlation coefficient was 0.997 (i pc = −55.74v − 0.590) and 0.996 (i pa = 59.15v + 4.799), respectively. This phenomenon suggests that the redox process is adsorption-controlled and the immobilized tyrosinase was stable. However, there is clearly a systematic deviation from linearity in this data set (i.e. low scan rates are always on one side of the line and the high scan rate points are on the other). The anodic and cathodic peak potentials are linearly dependent on the logarithm of scan rate (v), when v > 1.0 V s−1, which is in agreement with the Laviron theory, with slopes of −2.3RT/αnF and 2.3RT/(1 − α)nF for the cathodic and anodic peak, respectively. Subsequently, the charge-transfer coefficient (α) was estimated as 0.41.
The heterogeneous electron transfer rate constant (k s) was estimated according to the following equation [43]:
Here, n is the number of transferred electrons of the rate determining reaction and R, T and F possess their conventional meanings. ΔE p is the peak potential separation. The heterogeneous electron transfer rate constant (k s) can be calculated as 1.15 ± 0.04 s−1.
The stability of immobilized tyrosinase on the nanometer-scale nickel oxide surfaces has been evaluated by voltammetry in Fig. 5. The peak potential of the Tyr/NiO NPs/GC electrode remains approximately unchanged during the continuous cyclic voltammograms between 0.5 and −0.6 V. The surface area under the 50th voltammogram was almost 1.8% smaller than that under the first voltammogram. This difference reduced to 0.7% for the 50th and 100th voltammograms at a scan rate of 120 mV s−1. This is likely due to the surface area under the voltammogram (indicating the consumed charge/Q) being related to the amount of tyrosinase enzyme in electrical contact with the NiO NP surfaces. Therefore, amount of the enzyme in electrical contact, after the 50th cycle, was about 98.2% of the initial value at first cycle, equivalent to 99.3% for the 50th to 100th cycles.

Continuous CVs of the Tyr/NiO NPs/GC electrode from outer to inner; 1st, 50th and 100th CV in PBS, scan rate: 120 mV s−1
Figure 6 demonstrates voltammograms for the fresh Tyr/NiO NPs/GC electrode (CV a) and Tyr/NiO NPs/GC retained electrode (CV b) in PBS (4–6 °C) for 18 h. The height of the C1 reductive peak is increased (Fig. 6, CV b). In addition, the ΔE value diminishes in the case of the retained electrode (CV b). This may be caused by either or both; (i) An increase in the amount of tyrosinase in electrical contact with the NiO NP surfaces (the connectivity of protein prosthetic groups with the NiO NPs could be affected by enzyme rearrangements on the NiO NP surfaces); and/or, (ii) Structural changes at the electrode/solution interface, e.g. changes to the nickel oxide structure during the continuous voltammograms and over the time period (18 h).

CVs of Tyr/NiO NPs/GC in PBS (a) fresh electrode (b) retained electrode in PBS (4–6 °C, 18 h, pH 7.0), at 50 mV s−1
Figure 7 shows the continuous CVs of the Tyr/NiO NPs/GC retained electrode. The anodic and cathodic peak currents remain stable with cycling. The voltammograms in Fig. 7 indicate the stability of immobilized tyrosinase on the Tyr/NiO NPs/GC retained electrode.

The continuous CVs of the Tyr/NiO NPs/GC retained electrode in PBS (pH 7.0, 0.05 M), scan rate; 50 mV s−1
It is known that tyrosinase is a bifunctional enzyme, which catalysis o-diphenols to corresponding quinones [44] also referred to as diphenolase activity. Thus, this experiment examined the process of epinephrine catalysis to its corresponding quinone using immobilized tyrosinase on the Tyr/NiO NPs/GC electrode.
The ability of the Tyr/NiO NPs/GC electrode for epinephrine catalysis has been assessed by differential pulse voltammetry (DPV).
Figure 8 shows DPVs of the Tyr/NiO NPs/GC electrode in the absence and presence of epinephrine. As is observed in Fig. 8, the oxidation current of the electrode was greatly increased due to catalytic oxidation of epinephrine by the immobilized tyrosinase on Tyr/NiO NPs/GC electrode. Clearly, when epinephrine was added to the buffer solution, the DPV behavior of the electrode changed considerably. The oxidation peak current increased with epinephrine concentration increase in the solution. The increases in oxidative peak current confirmed that the tyrosinase enzyme on the Tyr/NiO NPs/GC electrode illustrated a catalytic ability to accomplish epinephrine oxidation.

Differential pulse voltammograms corresponding to the electrochemical responses of Tyr/NiO NPs/GC electrode generated by different epinephrine concentrations in PBS from inner to outer; 0, 0.5, 0.7, 1.0 and 3.0 μM. The step potential and modulation amplitude values of 5 and 25 mV, respectively
4 Conclusions
Nickel oxide nanoparticles (NiO NPs) were electrodeposited onto the surface of GC electrode and assessed using SEM and AFM procedures. We have demonstrated that the tyrosinase was immobilized onto the surface of NiO NPs and the resulting Tyr/NiO NPs were successfully applied to a glassy carbon substrate. The direct electrochemistry of tyrosinase in the form of a Tyr/NiO NPs/GC electrode was assessed by cyclic voltammetry.
References
Daniel M-C, Astruc D (2004) Chem Rev 104:293
Wanekaya AK, Chen W, Myung NV, Mulchandai A (2006) Electroanalysis 18:533
Niemeyer CM (2001) Angew Chem Int Ed 40:4128
Katz E, Willner I (2004) Angew Chem Int Ed 43:6042
Baron R, Lioubashevski O, Katz E, Niazov T, Willner I (2006) Angew Chem Int Ed 45:1572
Willner I, Baron R, Willner B (2006) Adv Mater 18:1109
Hallet B, Sherratt DJ (1997) FEMS Microbiol Rev 21:157
Wu L-Q, Payne GF (2004) Trends Biotechnol 22:593
Xu Y, Stokes AH, Freeman WM, Kumer SC, Vogt BA, Vrana KE (1997) Mol Brain Res 45:159
Kounalakis N, Goydos JS (2005) Curr Oncol Rep 7:377
Yaropolov AI, Kharybin AN, Emneus J, Marko-Varga G, Gorton L (1996) Bioelectrochem Bioenerg 40:49
Ye B, Zhou X (1997) Talanta 44:831
Dempsey E, Diamond D, Collier A (2004) Biosens Bioelectron 20:367
Sanz VC, Mena ML, Gonzalez-Cortes A, Yanez-Sedeno P, Pingarron JM (2005) Anal Chim Acta 528:1
Wang J, Lu F, Lopez D (1994) Analyst 119:455
Campuzano S, Serra B, Pedrero M, Villena FJM, Pingarron JM (2003) Anal Chim Acta 494:187
Zhou YL, Tian RH, Zhi JF (2007) Biosens Bioelectron 22:822
Liu Z, Liu Y, Yang H, Yang Y, Shen G, Yu R (2005) Anal Chim Acta 533:3
Li Y-F, Liu Z-M, Liu Y-L, Yang Y-H, Shen G-L, Yu R-Q (2006) Anal Biochem 349:33
Liu S, Yu J, Ju H (2003) J Electroanal Chem 540:61
Rogers KR, Becker JY, Cembrano J (2000) Electrochim Acta 45:4373
Burestedt E, Narvaez A, Ruzgas T, Gorton L, Emneus J, Dominguez E, Marko-Varga G (1996) Anal Chem 68:1605
Daigle F, Leech D (1997) Anal Chem 69:4108
Coche-Guerente L, Desprez V, Labbe P (1999) J Electroanal Chem 470:61
Forzani ES, Rivas GA, Solis VM (1999) J Electroanal Chem 461:174
Conway BE, Sattar MA (1968) J Electroanal Chem 19:351
Fleischmann M, Korinek K, Pletcher D (1971) J Electroanal Chem 31:39
Cowan RL, Staehle RW (1971) J Electrochem Soc 118:557
Giovanelli D, Lawrence NS, Jiang L, Jones TGJ, Compton RG (2003) Analyst 128:173
Wong JL, Tian M, Jin WR, He YN (2001) Electroanalysis 12:1355
Giovanelli D, Lawrence NS, Wilkins SJ, Jiang L, Jones TGJ, Compton RG (2003) Talanta 61:211
Salimi A, Sharifi E, Noorbakhsh A, Soltanian S (2006) Electrochem Commun 8:1499
Salimi A, Sharifi E, Noorbakhsh A, Soltanian S (2007) Biophys Chem 125:540
Bayandori Moghaddam A, Ganjali MR, Dinarvand R, Razavi T, Saboury AA, Moosavi-Movahedi AA, Norouzi P (2008) J Electroanal Chem 614:83
Bayandori Moghaddam A, Ganjali MR, Dinarvand R, Saboury AA, Razavi T, Moosavi-Movahedi AA, Norouzi P (2007) Biophys Chem 129:259
Mu C, Zhao Q, Xu D, Zhuang Q, Shao Y (2007) J Phys Chem B 111:1491
Bayandori Moghaddam A, Kobarfard F, Fakhari AR, Nematollahi D, Hosseiny Davarani SS (2005) Electrochim Acta 51:739
Bayandori Moghaddam A, Ganjali MR, Dinarvand R, Norouzi P, Saboury AA, Moosavi-Movahedi AA (2007) Biophys Chem 128:30
Bayandori Moghaddam A, Ganjali MR, Norouzi P, Niasari M (2007) J Electroanal Chem 601:205
Ottenbrite RM, Chiellini E (1992) Polymers in medicine: biomedical and pharmaceutical applications. PA, Technomic Publishing, Lancaster
Makino N, McMahill P, Mason HS (1974) J Biol Chem 249:6062
Ros-Martinez JR, Rodriguez-Lopez JN, Castellanos RV, Carsia Canovas F (1993) Biochem J 294:621
Laviron E (1979) J Electroanal Chem 101:19
Lerch K (1983) Mol Cell Biochem 52:125
Acknowledgement
Financial support from the University of Tehran Research Affairs is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bayandori Moghaddam, A., Ganjali, M.R., Saboury, A.A. et al. Electrodeposition of nickel oxide nanoparticles on glassy carbon surfaces: application to the direct electron transfer of tyrosinase. J Appl Electrochem 38, 1233–1239 (2008). https://doi.org/10.1007/s10800-008-9541-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10800-008-9541-3