
Abstract
In recent years, there has been increasing interest in studying the anti-inflammatory activity of polyunsaturated fatty acid ethanolamides (N-acylethanolamines, NAE), which are highly active lipid mediators. The results of this study demonstrate that a dietary supplement (DS) of fatty acid-derived NAEs reduces LPS-induced inflammation. The processes of cell proliferation, as well as the dynamics of Iba-1-, CD68-, and CD163-positive macrophage activity within the thymus and spleen were studied. The production of pro-inflammatory cytokines (TNF, IL1β, IL6, and INFγ), ROS, NO, and nitrites was evaluated in the blood serum, thymus, and LPS-stimulated RAW264.7 mouse macrophages. In vitro and in vivo experiments have shown that DS (1) prevents LPS-induced changes in the morphological structure of the thymus and spleen; (2) levels out changes in cell proliferation; (3) inhibits the activity of Iba-1 and CD68-positive cells; (4) reduces the production of pro-inflammatory cytokines (TNF, IL1β, IL6, and INFγ), ROS, and CD68; and (5) enhances the activity of CD-163-positive cells. In general, the results of this study demonstrate the complex effect of DS on inflammatory processes in the central and peripheral immune systems.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
The body’s physiological response to various exogenous or endogenous stimuli, such as toxins, cell damage, or pathogens, is inflammation [29]. The goal of the inflammatory response is to localize and eliminate damaging factors, initiating the repair process and further recovery [10, 21, 35]. Therefore, the inflammatory process is regulated by a wide range of signaling molecules that induce, maintain, and stop it. An imbalance between the production of signaling molecules leads to uncontrolled inflammation, taking part in the pathogenesis of autoimmune diseases [14, 15]. For example, in rheumatoid arthritis (RA), macrophages contribute significantly to inflammation and joint destruction. During RA, the most common cell type in the synovial membrane is macrophages polarized into the M1 phenotype [28].
Macrophages are central to the innate immune system and are involved in the control of the inflammatory response [1]. They can secrete trophic factors, immune mediators, and effectors, phagocytize, and present antigens on the cell surface [17]. Macrophages are characterized by plasticity and diversity of phenotypes and functions, which are influenced by both their origin and the microenvironment of the resident tissue [19, 30]. They polarize in two different subtypes—classically (M1-type) and alternatively (M2-type) activated macrophages [1, 29].
M1 macrophages exhibit strong antimicrobial properties, enhance Th1 responses, and exert antitumor activity. Induce the polarization of macrophages M1-type pathogens or pro-inflammatory cytokines, such as tumor necrosis factor (TNF), interferon-γ (IFNγ), and granulocyte macrophage colony-stimulating factor (GM-CSF). All this leads to the activation of a number of signaling events that enhance the production of mediators [9, 11]. They express pro-inflammatory cytokines (IL1β, IL6, IL12, IL23, and TNF), reactive oxygen species (ROS), and nitric oxide (NO). In addition, a key aspect of macrophage polarization is a change in the expression of cell surface markers. M1 macrophages express high levels of major histocompatibility complex class II (MHC-II), marker CD68, and costimulatory molecules CD80 and CD86 [9, 18, 19, 32]. Lipopolysaccharide (LPS) is used in experiments to polarize M1 macrophages. Their activation occurs through TLR4, TRIF, and MyD88. TRIF-regulated mechanism triggers kinase cascades and ultimately activates IRF3, and secretion of IFN, including IFNα and IFNβ, occurs. MyD88, which is another adapter, activates the nuclear factor kappa-B (NF-kB) pathway (p65 and p50), a critical transcription factor in M1 macrophage polarization. MyD88 also activates AP-1 via MAPK. These pathways promote the expression of various inflammatory genes such as pro-inflammatory cytokines (including tumor necrosis factors (TNF), IL1β, and IL12), chemokines CXCL10, CXCL11, costimulatory proteins, and proteins that process antigens [9, 11]. Thus, M1 macrophages produce pro-inflammatory cytokines and are involved in the formation of the inflammatory response. However, their hyperactivation can lead to tissue damage, which is harmful to health [12, 55].
In contrast, M2-type macrophages play a central role in parasite responses, tissue remodeling, angiogenesis, and allergic diseases. Phenotypically, this population is characterized by the expression of the mannose receptor CD206 (MRC1), membrane glycoprotein CD200R, CD163 [9, 46]. They are polarized by IL4 and IL13 and promote the Th2 response, as well as produce anti-inflammatory cytokines—IL10 and transforming growth factor beta (TGF-β) [19, 30]. M2-type macrophages are divided into M2a (secrete TGFβ, IL10), M2b (secrete TNF, IL1, IL6, IL10), M2c (secrete IL10, CD206, TGFβ), and M2d (secrete IL10, IL12, VEGF, TNF) [26]. Thus, macrophages produce various biologically active molecules that can negatively and positively influence the inflammatory process. The search and development of new macrophage-based drugs and methods open up new opportunities for the control and treatment of various inflammatory diseases [15, 54].
In recent years, the use of polyunsaturated fatty acids (PUFAs) has attracted particular interest in various inflammatory diseases [6, 7, 48]. Among PUFAs, eicosapentaenoic and docosahexaenoic acids (EPA and DHA) are of particular attention due to their anti-inflammatory, provided by inhibition pro-inflammatory cytokines (TNF, IL1β, IL6), reducing eicosanoids and leukocyte chemotaxis involved in inflammation [6, 20]. PUFA derivatives N-acylethanolamines (NAE) are also involved in inflammatory processes, but their influence is poorly understood. PUFA ethanolamides are known to act as biologically active lipid messengers and play an important role in the inflammatory process [39].
N-docosahexaenoylethanolamine (DHEA) and N-eicosapentaenoylethanolamine (EPEA) are considered the most studied and effective PUFA ethanolamides. Both EPEA and DHEA have anti-inflammatory properties, induce apoptosis, and have anti-proliferative effects on certain tumor cell lines. DHEA and EPEA have been shown to have an inhibitory effect on the growth of human prostate cancer cells LNCaP and PC3 [5], as well as the induction of autophagy in cultured human breast cancer cells MCF-7 [45]. When modeling LPS-induced inflammation in macrophage cell culture in vitro, treatment with EPEA and DHEA reduced the synthesis of the pro-inflammatory chemokine MPC-1 in a dose-dependent manner [36]. It has also been found that the anti-inflammatory effect of DHEA on macrophages is predominantly due to the inhibition of eicosanoids produced by COX-2 [38]. It is worth noting that nanomolar concentrations of DHEA promotes neurogenesis, neuronal growth, and synaptogenesis in developing neurons. Park et al. [40] found that DHEA suppressed LPS-induced neuroinflammation by enhancing cAMP/PKA signaling and inhibiting NF-κB activation. DHEA has also been shown to bind to GPR110 and induce cAMP production and phosphorylation of protein kinase A (PKA) and cAMP response element-binding protein (CREB). This signaling pathway results in downregulation of pro-inflammatory gene expression [24]. In vivo, DHEA and EPEA prevented LPS-mediated astrogliosis and also polarized microglia towards the M2 type [51]. Our previous study demonstrated the anti-inflammatory activity of a dietary supplement (DS) based on NAE fatty acids. DS suppressed the neuroinflammatory response by reducing glia activation, positively regulated the proliferation of neuronal progenitors, and attenuated the hippocampus-dependent memory impairment that developed with LPS administration [52].
The present study is aimed at a detailed evaluation of the DS effects in the central and peripheral immune systems during LPS-induced inflammation. The main components of DS are N-docosahexaenoylethanolamine (DHEA), N-eicosapentanoylethanolamine (EPEA), N-stearoylethanolamine (SEA), the content of which is 29.81%, 27.04%, and 12.42%, respectively [52].
MATERIALS AND METHODS
N-Acylethanolamine Preparation
The Latyshev [27] method was used to isolate the concentrate of polyunsaturated fatty acids (PUFA). The source of PUFA was the squid Berryteuthis magister fished in the western part of the Bering Sea at a depth of 400–600 m. Mass spectra of methyltrisilyl derivatives of PUFA ethanolamides obtained by GC–MS were published in our previous article [52].
Cell Culture
For in vitro studies, the RAW264.7 mouse macrophage cell line provided by the American Type Culture Collection was used. The standard culture medium consisted of DMEM/F12 (Thermo Fisher Scientific, Waltham, Massachusetts, USA) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, Waltham, Massachusetts, USA), essential amino acid solution (Gibco, USA), and penicillin–streptomycin (Thermo Fisher Scientific, Waltham, MA, USA). An incubator (MCO-18AIC, Sanyo, Osaka, Japan) was used, set to 5% CO2, and 37 °C. To study the anti-inflammatory activity, cells previously seeded into a 96-well microplate (1 × 103 cells/well) were treated with DS at concentrations of 0.1, 1, and 10 µg/ml for 1 h and then activated by lipopolysaccharide (LPS, E. coli O111:B4, Sigma-Aldrich, Bellefonte, PA, USA) at a concentration of 1 µg/ml. Cells cultured in a culture medium containing only LPS were used as a positive control.
MTS Test
The cytotoxic effect of DS (at concentrations of 0.1, 1, and 10 μg/ml) was studied by viability analysis using tetrazolium compound MTS. After incubation with DS, the medium in all wells was carefully removed and replaced with fresh medium with the addition of 1/10 of the volume of the MTS reagent (Abcam, ab197010, USA). The plate was incubated under standard conditions for 2 h. The iMark microplate reader was used to measure the absorbance (490 nm) (Bio-Rad, USA). Cell viability was calculated setting absorbance in control (untreated) cells at 100% and absorbance in empty wells without cells at 0%.
Analysis of Reactive Oxygen Species (ROS) and Nitric Oxide (NO)
ROS activity was studied after a 24-h incubation of macrophage cells with LPS and DS. Following manufacturer’s recommendation, 20 µl of 2,7-dichlorodihydrofluorescein diacetate solution (D399, Invitrogen) was supplemented, and the microplate was incubated for 10 min at 37 °C. To analyze the NO level, a 10 μM DAF-FM diacetate solution (D23844, Thermo Fisher Scientific, USA) was used in accordance with the recommendations of manufacturer. Fluorescence intensity was measured using a SPARK TECAN 10 M plate reader for ROS at λex/λem = 485/518 nm and for NO λex/λem = 460/524 nm. The results obtained are presented as percentages relative to the negative control.
Nitrite Quantification
After cell incubation with LPS and DS, nitrites were measured by the Griess method. Griess reagent (100 µl) (Sigma, G4410, CШA) were added to 100 µl of cell culture medium and brooded for 10 min at RT. Used an iMark microplate reader to measure the absorbance (540 nm) (Bio-Rad, USA).
Animals
The experimental animals were kept under vivarium conditions at constant temperature (23 °C ± 2 °C) and humidity (55% ± 15%). Male mice (3 months old, strain C57BL/6) were housed 8 mice per cage with free access to food and water and a 12-h light/dark cycle. All experimental procedures were approved by the Animal Ethics Committee at the A.V. Zhirmunsky National Scientific Center of Marine Biology, Far Eastern Branch, Russian Academy of Sciences (No. 1.3/2022), according to the Laboratory Animal Welfare guidelines and the European Communities Council Directive 2010/63/EU. To form a model of LPS-induced inflammation, mice were intraperitoneally injected with a solution of lipopolysaccharides (LPS, Escherichia coli O111:B4, Sigma-Aldrich, St. Louis, MO, USA) at a dose of 700 μg/kg 2 times with an interval of 24 h. DS was administered orally via gavage at a dose of 10 mg/kg daily for seven days. All mice were divided into 4 groups: “Veh” (n = 16)—i.p. saline injection and oral water administration; “Veh + DS” (n = 16)—i.p. saline and oral DS administration; “LPS” (n = 16)—i.p. injection of LPS and oral water administration; “LPS + DS” (n = 16)—i.p. LPS injected and oral DS administration.
ELISA
To quantify the concentration of TNF, IL1β, IL6, and INFγ in the serum and thymus of mice, an ELISA was performed. Mice (n = 8 animals/group) were anesthetized (3% isoflurane in 100% oxygen); then, the chest cavity was opened, and blood was drawn from the left ventricle using a syringe. Then, each blood sample was transferred to an individual serum tube (Greiner Bio One, Kremsmünster, Austria), coated with a clotting activator, and centrifuged. Then, the blood serum separated from the formed elements was transferred into individual test tubes and frozen in liquid nitrogen. After that, the thymus tissue was removed and frozen in liquid nitrogen. Thymus samples were homogenized in ice-cold extraction buffer (100 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EGTA, 1 mM EDTA, 1% Triton X-100, and 0.5% sodium deoxycholate) supplemented with a cocktail of protease inhibitors 1 mg/ml (Complete; Sigma-Aldrich, St. Louis, MO, USA) and phosphatase inhibitor mixture 0.01 mg/ml (P5726, Sigma-Aldrich, St. Louis, MO, USA). Serum samples were simply diluted with extraction buffer without homogenization. Protein concentration was determined using a BCA protein assay kit (Pierce, Rockford, IL, USA). Samples were mixed with bicarbonate-carbonate buffer (100 M, 3.03 g Na2CO3, 6.0 g NaHCO3, and 1000 ml distilled water, pH 9.6) to give a final concentration of 20 μg/ml. The sample was dispensed at 100 µl into a PVC microtiter plate (M4561-40 EA, Greiner, Kremsmünster, Austria) and incubated overnight at + 4 °C. After washing 3 times, 200 µl of buffer (PBS) was added to each well. Five percent skimmed milk powder (M7409-1BTL, Sigma-Aldrich, St. Louis, MO, USA) (2 h at RT) was used to block the remaining protein-binding sites in the coated wells. After washing, a solution of primary antibodies in a volume of 100 µl was added to the samples. The following primary antibodies were used: TNF (1:1000, ab208348), IL1β (1:1000, ab205924), IL6 (1:500, ab46100), and INFγ (1:1000, ab216642), all from Abcam (Cambridge, UK). The plate was incubated at RT in a solution of primary antibodies for 2 h. The solutions were then changed in shifts, washing three times with PBS after each step: 100 µl of secondary anti-rabbit antibody solution (1:500, PI-1000, Vector Laboratories, San Francisco, CA, USA) (2 h, RT); 50 µl TMB (3,3′,5,5′-tetramethylbenzidine, SK-4400, Vector Laboratories, San Francisco, CA, USA) solution (RT, 30 min until color appears); and 50 µl stop solution (1 N hydrochloric acid). Each plate was analyzed twice on an iMark microplate reader (Bio-Rad, USA) at a wavelength of 450 nm.
Western Blot
Western blot was used to detect TNF, IL1β, IL6, and CD68 levels in RAW264.7 mouse macrophage and mice serum levels TNF, IL1β, and IL6. RAW264.7 cell lysates were sonicated with 0.150 mM serine protease inhibitor (PMSF) solution prior to protein measurement and adjustment to 2 mg/ml. Serum samples were simply diluted with extraction buffer without homogenization. Loading buffer (sample buffer, Bio-Rad, Hercules, CA, USA) containing 5% 2-mercaptoethanol was mixed 1:1 with the samples and placed in a water bath (94.5 °C for 5 min). For electrophoresis, ready-made cartridges Protean mini gel Any kDa (Bio-Rad, Hercules, CA, USA) and Spectra Multicolor Broad Range Protein Ladder (Thermo Fisher Scientific, Waltham, MA, USA) were used in the Bio-Rad system (load per well 60 μg of protein, current per gel 15 mA). Transfer of proteins to the PVDF membrane was performed using the Turbo transblot transfer system (Bio-Rad, Hercules, CA, USA). Transblot Turbo RTA Transfer Kit (Bio-Rad, Hercules, CA, USA) was used for transfer. The membrane was then placed overnight at + 4 °C in blocking buffer (PBS containing 2% BSA, 0.1% Tween 20, 0.05% TritonX100). The next day, the blocking buffer was washed with PBS-T, after which it was incubated for 1 h with primary antibodies. The following primary antibodies were used: TNF (1:1000, ab208348), IL1β (1:1000, ab205924), IL6 (1:500, ab46100), and CD68 (1:1000, ab125212), all from Abcam (Cambridge, UK). Moreover, antibodies to α-Tubulin (1:1000, Sigma-Aldrich, T6199, St. Louis, MO, USA) and β-Actin (1:5000, Thermo Fisher Scientific, MA1-140, Waltham, MA, USA) were used as load control. After incubation with primary antibodies, the membranes were washed with PBS-T and then incubated for an hour with secondary antibodies (1:500, Anti Rabbit, PI-1000 and Anti Mouse, PI-2000, Vector Labs, San Francisco, CA, USA). The membrane was then coated with Western Blot ECL Substrate (Bio-Rad, Hercules, CA, USA) (1 ml substrate per membrane, 5 min). Visualization was performed using the ChemiDoc gel documentation system (Bio-Rad, Hercules, CA, USA). The resulting images were analyzed using the ImageLab software package.
Histological and Immunohistochemistry Staining
The spleen and thymus were removed for histological and immunohistochemical studies on the 7th day after the start of the experiment. For anesthesia, inhalation anesthetic 3% isoflurane in 100% oxygen (Laboratories Karizoo, S.A., Barcelona, Spain) was used. Anesthetized mice (n = 8 animals per group) were transcardially perfused with 5 ml 0.1 M PBS (4 °C), pH 7.2, followed by 5 ml cold fixative (4% paraformaldehyde in 0.1 M PBS, pH 7,2). Then, the spleen and thymus were rapidly removed and placed in 4% paraformaldehyde for 12 h. After washing in PBS, the spleen and thymus were embedded in paraffin. Using a Leica RM 2245 microtome (Leica, Wetzlar, Germany), sections were made (7 µm). Paraffin sections of the spleen and thymus were stained with hematoxylin–eosin.
For immunohistochemical study, sections after deparaffinization were incubated in 0.3% H2O2 for 5 min. A solution of 2% BSA and 0.01% Triton X100 in 0.1 M PBS (pH 7.2) was used to block nonspecific antibody binding. Further incubation was carried out with a solution of primary antibodies (overnight, 4 °C). For immunohistological studies, antibodies against Iba-1 (1:500, ab108539), CD68 (1:500, ab125212), CD163 (1:500, ab182422), and mouse monoclonal antibodies to PCNA (1:1000, ab29) were used. After washing with PBS-T, the sections were placed in a solution of secondary antibodies (1:100, Anti Rabbit, PI-1000 and Anti Mouse, PI-2000, Vector Laboratories, San Francisco, CA, USA). Staining was performed using DAB peroxidase chromogen substrate (Abcam, Cambridge, UK). After washing with distilled water, the sections were dehydrated and mounted under glass slides using mounting medium (CS705, Dako, Denver, Colorado, USA).
Visualization of Collected Data
Images were visualized using a Zeiss Axio Imager microscope equipped with an AxioCam 503 color camera and Axio-Vision software (Carl Zeiss, Oberkochen, Germany) and used ImageJ software for image processing and analysis (NIH, Bethesda, MD, USA). At least 80 images of the spleen and thymus for each group of animals were used for analysis. The count of PCNA-positive cells in the spleen (white pulp) was performed manually using the cell counter plugin. The number of PCNA-positive cells/mm3 was calculated by the formula: d = (109 × n)/(S × 7), where d is equal to the cell density; 109 is the coefficient converting µm2 to mm3; n is the number of immunopositive cells; S is the area of the white pulp follicle (µm2); and 7 is the slice thickness (µm). Evaluations of staining area specific for Iba-1-, CD68-, and CD163-positive cells were performed using every sixth section. For this, the obtained photographs were processed by converting the image to grayscale mode (16-bit image), background subtraction, and binarization. The area of immunopositive staining was expressed as a percentage. The operator performing the measurements was not aware of the identity of the sections.
Statistical Analysis
Data were statistically processed using one-way ANOVA followed by Tukey’s post hoc test. Using the Shapiro–Wilk test, all data were checked for normal distribution. The data obtained from the in vitro study were compared using Mann–Whitney test. Data were presented as mean ± SEM (standard error of the mean), and p < 0.05, p < 0.01, and p < 0.001 were considered statistically significant. All statistical tests and plotting were performed using GraphPad Prism 4.00 software (GraphPad Software, San Diego, CA, USA).
RESULTS
DS Administration Reduces the Production of Pro-inflammatory Cytokines In Vitro
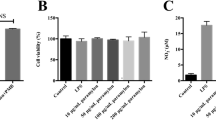
In our study, using the MTS test, the absence of the cytotoxic effect of DS at the studied concentrations on the RAW264.7 macrophage cell line was shown (Fig. 1a). The addition of LPS (1 µg/ml) to macrophages led to a significant increase in pro-inflammatory markers: reactive oxygen species (ROS), nitrites, and nitric oxide (NO). The addition of DS to the LPS-activated macrophages at the studied concentrations (0.1–10 µg/ml) resulted in a statistically significant decrease in ROS (p < 0.05, p < 0.01) (Fig. 1b). However, DS had no effect on NO (Fig. 1c) and nitrite (Fig. 1d) levels. Western blot data demonstrate increased production of TNF, IL1β, IL6, and CD68 in LPS-activated macrophages. The addition of DS at concentrations of 1 and 10 µg/ml reduced the production of pro-inflammatory markers almost to control values (Fig. 1e, f).

DS biological activity study using a macrophage cell line RAW264.7. a Effect of DS on macrophage viability and production of b ROS, c NO, and d nitrite. Data are the mean ± SEM, n = 9 (number of analyzed samples), ***p < 0.001—compared to “Veh”; +p < 0.05, ++p < 0.01, +++p < 0.001—compared to “LPS” (Mann–Whitney test). e Western blot analysis of TNF, IL1β, IL6, and CD68 levels in macrophages. f Relative quantity of TNF, IL1β, IL6, and CD68 in RAW264.7 macrophage lysates, data are the mean ± SEM, n = 4 (number of analyzed samples), *p < 0.01—compared to “Veh”, +p < 0.001—compared to “LPS” (Mann–Whitney test). Data normalized to α-Tubulin.
DS Administration Decreases Production of Proinflammatory Cytokines in Serum Mice
Using the ELISA method, it was found that inflammation caused by LPS led to a 1.5–twofold increase in the synthesis of proinflammatory cytokines (IL1β, TNF, and IL6) in the blood serum of experimental animals. DS administration almost completely prevented the LPS-induced increase in serum pro-inflammatory cytokine synthesis (Fig. 2a). Data from Western blot analysis additionally confirm that LPS exposure also increased the concentration of pro-inflammatory cytokines in the blood serum of mice from the LPS group. The use of DS led to a decrease in the production of TNF, IL1β, and IL6 to the values of the “Veh” and “Veh + DS” groups (Fig. 2b, c).

The effect of LPS and DS administration on the activation of cytokines in the serum of mice. a ELISA level analysis of TNF, IL1β, and IL6 expression in the serum, data are the mean ± SEM, n = 8 (number of analyzed samples), *p < 0.05, **p < 0.01, ***p < 0.001 (one-way ANOVA). b Western blot analysis of TNF, IL1β, and IL6 in the serum. c Relative quantity of TNF, IL1β, and IL6 in the serum, data are the mean ± SEM, n = 4 (number of analyzed samples), *p < 0.05, **p < 0.01, ***p < 0.001 (one-way ANOVA). Data normalized to β-Actin.
DS Administration Prevented the Development of Morphological Changes and Reduced Proliferative Activity in the Thymus and Spleen of Mice
The thymus and spleen of animals from the “Veh” group had a typical histological structure: the thymus is externally covered with a connective tissue capsule; partitions extending from it divide the parenchyma into lobules of various sizes. The lobules are composed of cortical and medulla zone. The spleen tissue can be divided into red and white pulp, which are separated from each other by a marginal zone. The red pulp is located between the white pulp and consists of reticular tissue penetrated by venous sinuses. The white pulp of the spleen is formed by periarterial lymphoid muffs, consisting of lymphoid tissue surrounding the central arteries (Fig. 3a). DS administration in “Veh + DS” mice did not change the white/red pulp area ratio (W/R index), which was maintained at the “Veh” group level (0.7 ± 0.03 and 0.7 ± 0.03, respectively). LPS administration led to the development of an inflammatory reaction. The boundaries between the cortex and medulla in the thymus lobules became less distinguishable, and the density in the lymphocytes’ arrangement changed. Within the spleen, there was an increase in white pulp follicles, which was accompanied by a change in the ratio of the W/R index (1.9 ± 0.1). In LPS-treated mice, DS administration prevented the development of morphological changes typical for LPS-induced inflammation within the thymus and spleen. In the thymus lobules, clearer boundaries between the cortical and medulla were observed. In addition, there was a decrease in the size of the white pulp follicles (0.6 ± 0.02) compared to the level of the “Veh” groups within the spleen (Fig. 3a, c).

Sections of the mice thymus and spleen, a stained with H&E (C, cortex; M, medulla; R, red pulp; W, white pulp). b Distribution of PCNA-positive cells (black arrows). c The area ratio of white and red pulp in the spleen. d PCNA-positive cells in the thymus and e white pulp of the spleen. Data are the mean ± SEM, n = 8/group, *p < 0.05, **p < 0.01, and ***p < 0.001 (one-way ANOVA). Scale bar—50 µm.
The most common way to determine the proliferative activity of cells is the use of immunohistochemical methods, in particular, antibodies to the PCNA marker (proliferating cell nuclear antigen). In our work, in the thymus, the number of PCNA-positive cells in the “Veh” and “Veh + DS” groups were 43,130 ± 3424 cells/mm3 and 34,240 ± 1800 cells/mm3, respectively. In addition, in the spleen of the same groups, the number of PCNA-positive cells was 94,420 ± 2826 cells/mm3 and 81,120 ± 4083 cells/mm3, respectively. The number of PCNA-positive cells in the thymus (76,360 ± 2229 cells/mm3) and white pulp of the spleen (129,100 ± 5784 cells/mm3) increased after LPS administration. At the same time, in the “LPS + DS” group, the opposite effect was observed. The proliferative activity of cells within the thymus and follicles of the spleen white pulp decreased to the level of the control groups (Fig. 3b, d, e).
DS Administration Reduced the Development of an Inflammatory Response in the Thymus and Spleen After LPS Administration
In this work, we studied the effect of DS on the production of pro-inflammatory (CD68) and anti-inflammatory macrophage markers (CD163) within the thymus and spleen of mice. Statistical differences in the thymus were not found between the “Veh” and “Veh + DS” groups when assessing the staining area of Iba-1-positive cells (18 ± 1.2% and 17 ± 0.9%, respectively). The LPS administration resulted in the Iba-1-positive staining area increase (48 ± 2.6%), while DS administration was accompanied by its decrease almost to the values of the “Veh” group (26.8 ± 1.6%) (Figs. 4a and 5b). In the white pulp of the spleen, the LPS treatment did not cause an Iba-1-positive staining area increase (21.9 ± 1%) in comparison with the control groups. DS administration in LPS-treated mice caused a decrease in the Iba-1-positive area (10.3 ± 0.8%) compared to other experimental groups. The opposite result was obtained during the study of the red pulp of the spleen. The LPS administration led to an increase in the Iba-1-positive area (30.4 ± 1.8%) compared to the “Veh” and “Veh + DS” groups (23.6 ± 1% and 23.1 ± 0.9%, respectively). DS administration prevented the development of an inflammatory response and reduced the Iba-1-positive cells’ staining area below the level of control groups (12.7 ± 1.3%) (Figs. 4a and 5e).

Immunohistochemical staining of a Iba-1-, b CD68-, and c CD163-positive cells within the thymus and spleen. Scale bar—50 µm.

The effect of LPS and DS administration on pro-inflammatory and macrophage markers level within the mice thymus and spleen. a The level of TNF, IL1β, IL6, and INFγ expression in the thymus measured by ELISA. Area staining of Iba-1-positive cells in the b thymus and e spleen. Area staining of CD68-positive cells in the c thymus and f spleen. Area staining of CD163-positive cells in the d thymus and g the red pulp of spleen. Data are the mean ± SEM, n = 8/group, *p < 0.05, **p < 0.01, and ***p < 0.001 (one-way ANOVA).
Anti-CD68 antibodies were used to assess the activity of pro-inflammatory macrophages. In the “Veh” and “Veh + DS” groups, there were no statistically significant differences in the CD68-positive cells’ staining area (2.1 ± 0.1% and 2.5 ± 0.1%, respectively). The immunopositive area increased after LPS administration to 3.7 ± 0.1%, while DS reduced the area to the level of control values (2.5 ± 0.1%) (Figs. 4b and 5c). The data obtained during immunohistochemical staining were additionally confirmed by ELISA; in the “LPS + DS” group, compared with “LPS,” there was a statistically significant decrease in pro-inflammatory cytokines, such as IL1β, IL6, and INFγ (Fig. 5a). The study of pro-inflammatory macrophages in the spleen showed that the CD68-positive cell distribution in the white and red pulp is not uniform. In the white pulp, there were no significant differences in the distribution of CD68-positive cells in the “Veh,” “Veh + DS,” and “LPS” groups (0.7 ± 0.06%, 0.6 ± 0.05%, and 0.7 ± 0.03%, respectively). However, a decrease in macrophage activity was observed in the “LPS + DS” group (0.5 ± 0.4%). In contrast to the white pulp, in the red pulp of the spleen, LPS significantly increased macrophage activity (3.5 ± 0.1%) compared to the “Veh” and “Veh + DS” groups (2.5 ± 0.2% and 2.3 ± 0.2%). The DS administration in LPS-treated mice reduced the staining area of CD68-positive cells below the level of control groups (1.6 ± 0.1%) (Figs. 4b and 5f).
The evaluation of anti-inflammatory macrophages using antibodies to CD163 showed that the staining area of CD163-positive cells in the thymus of the “Veh,” “Veh + DS,” and “LPS” animals did not statistically differ from each other (0.3 ± 0.03%, 0.3 ± 0.02%, and 0.5 ± 0.05%, respectively). At the same time, an increase in the CD163-positive cells staining area was observed in the “LPS + DS” group (0.8 ± 0.09%) (Figs. 4c and 5d). In the red pulp of the spleen, the LPS treatment led to a decrease in the staining area of CD163-positive cells to 1.1 ± 0.05%, compared to the “Veh” and “Veh + DS” groups (2.2 ± 0.1% and 2.3 ± 0.1%, respectively). The DS administration for 7 days in LPS-treated animals was accompanied by the CD163-positive staining area recovery (1.6 ± 0.1%). In the white pulp of the spleen, CD163-positive cells were almost not detected (Figs. 4c and 5g).
DISCUSSION
Monocytes and macrophages play one of the most important roles in the initiation and maintenance of chronic inflammation. They represent a heterogeneous population of cells and have a wide spectrum of activity, which allows them to modify their phenotype and functional properties depending on environmental conditions [1]. The formation of two different functional states of macrophages (M1/M2) is called “polarization” [2, 8]. M1-type are classically activated macrophages, the activation of which is observed under the action of LPS/IFNγ and is mediated by a large amount of NO release, proinflammatory cytokines, such as IL1α, IL1β, TNF, and IL6, and expression of CD68 and CD86 receptors. M1-type macrophages have pronounced antimicrobial and antitumor activity. A chronic inflammatory response leads to tissue damage due to the excessive release of ROS, which prevents repair processes [2, 16]. With the development of chronic inflammation, mechanisms are triggered that inhibit inflammation due to the activation of the M2-type anti-inflammatory macrophages. Polarization of the M2 type of macrophages occurs under the influence of IL4, IL13, and IL10 due to the activation of STAT6 and STAT3 signaling pathways [33, 42] and is characterized by increased release of IL10, TGFβ, arginase-1 (Arg1), CCL17, and CCL24 and expression of CD163 and CD206 receptors. M2-type macrophages provide the process of apoptotic cell phagocytosis, promote tissue repair and wound healing, have proangiogenic and profibrotic properties [23], reduce inflammation, and stimulate the remodeling of damaged tissues [22, 32]. However, to date, it has been shown that macrophage activation cannot be simply subdivided into M1 and M2 types. Macrophages, depending on the involved cytokine agonists and toll-like receptors, can exhibit many different forms of activation, derived from the two extreme states M1/M2. Pharmacological correction of the macrophages’ morphological and functional state is a promising strategy for anti-inflammatory therapy of a wide range of pathological processes (infectious intoxication, rheumatoid arthritis, atherosclerosis, etc.) [3, 11, 56].
The present study is focused on the anti-inflammatory properties of DS in the central and peripheral immune systems during the development of inflammation. The main components of DS are the omega-3 PUFA N-acylethanolamines (DHEA, EPEA, and SEA), which are part of a group of highly active anti-inflammatory lipid mediators. To induce inflammation in vitro and in vivo, we used Escherichia coli lipopolysaccharide (LPS), which is part of the gram-negative bacteria cell walls. LPS leads to the development of a persistent inflammatory response, which is accompanied by macrophage activation towards the pro-inflammatory M1 type [31, 44, 47]. In our study, intraperitoneal LPS administration for 2 days contributed to the initiation of an inflammatory process, which was accompanied by the pro-inflammatory cytokine (TNF, IL1β, IL6, and INFγ) production increase in the blood serum and thymus of experimental animals. In addition, there was a more pronounced thymus involution, including narrowing of the cortical area and the absence of division into cortical and medulla. The number of PCNA-positive proliferating cells increased, and activation of pro-inflammatory macrophages was also observed, as evidenced by a significant increase in the staining area of CD68-positive thymus cells. It has been previously shown that LPS-induced inflammation causes activation, proliferation, and migration of T-lymphocytes from the thymus to the peripheral organs of the immune system, in particular to the spleen [13]. Morphological changes caused by the LPS treatment were also observed in the spleen. During antigenic stimulation, an increase in the proliferative activity of cells was noted, accompanied by a change in the ratio of the area of the white pulp compared to the red one (W/R index). It should be noted that an increase in the CD68-positive staining area of M1-type macrophages was observed only in the red pulp of the spleen. This phenomenon may be because the red pulp is permeated with venous sinuses, in which the blood flow rate is reduced, resulting in more “close” and prolonged contact of antigens with macrophages [4]. The observed decrease in CD163-positive M2-type macrophages staining area in the red pulp of the spleen is probably associated with probably due to the fact that CD163 is a marker of M2 macrophages, and LPS inhibits the expression of CD163 mRNA and protein [25]. Oral DS administration in LPS-treated animals inhibited the production of IL1β, TNF, IL6, and INFγ in the serum and thymus of mice. A pronounced anti-inflammatory effect of DS was observed in the thymus and spleen, and the staining area of CD68-positive macrophages decreased. However, it is not entirely clear what consequences the observed decrease in the activity of the M1-type macrophages in the spleen will have for the body. At the same time, there was an increase in the CD163-positive macrophage staining area both in the thymus and in the spleen. In LPS-induced inflammation CD163 acts as a receptor involved in the clearance of hemoglobin/haptoglobin complexes by macrophages and may thus help to protect tissues from oxidative damage [43]. In addition, the DS administration during the development of inflammation contributed to the thymus and spleen morphological structure recovery. The W/R index and proliferative activity remained at the level of the control groups. It should be noted that the PCNA immunohistochemical marker used in our study is not only a marker of cell proliferation but also of cell repair. Therefore, we suggest that a decrease in PCNA level may indirectly indicate the pro-apoptotic activity inhibition in the thymus and spleen of mice. This effect of DS is probably realized through the regulation of anti-apoptotic proteins Bcl-2 and Bcl-xL, as well as inhibition of the pro-apoptotic proteins Bax and Bad expression, which was previously shown in the study of docosahexaenoic acid [41].
The results of our in vivo studies are also confirmed in vitro on the RAW264.7 macrophage cell line. Pretreatment of DS cells prevented the LPS-induced increase in the production of ROS, TNF, IL1β, and IL6, as well as the M1-type macrophage marker CD68. The pronounced anti-inflammatory effect of DS in in vivo and in vitro experiments is probably mediated by the activity of its main components (DHEA, EPEA, and SEA). Early other studies have shown that certain NAEs are able to reduce LPS-induced inflammation [40, 51]. However, almost all studies have focused on the study of neuroinflammation processes, while our data significantly expand the range of observed effects in the central and peripheral immune systems. Previously, in vitro experiments have shown that DHEA reduces the LPS-induced inflammatory response by inhibiting nuclear translocation, NF-κB promoter activity, and phosphorylation of mitogen-activated protein kinases (MAPKs) [40, 49]. The anti-inflammatory activity of some NAEs is mediated by a specific G-protein-coupled receptor GPR110 (ADGRF1). Some NAEs can interact with GPR110 to trigger cAMP production, phosphorylation of protein kinase A (PKA), and cAMP-binding protein (CREB) [24]. In addition, given that the bulk of NAE entering the body is hydrolyzed to individual PUFAs and ethanolamine by N-acylethanolamine-hydrolyzing acid amidase (NAAA) and fatty acid amide hydrolase (FAAH) [34, 50, 53], it cannot be ruled out that PUFAs also have some anti-inflammatory effect. Moreover, increasing the concentration of NAE in the body can suppress the activity of NAAA and FAAH concerning other endogenous NAE due to competitive inhibition, which also contributes to the resolution of the pathological condition [37].
According to our study, DS administration contributed to the reduction of the inflammatory process induced by LPS. Regulation of macrophage activity and inhibition of the pro-inflammatory molecules release probably underlie the DS anti-inflammatory activity and is mediated through several signaling pathways. The data obtained indicate a therapeutic potential of DS; however, the molecular mechanisms underlying its anti-inflammatory activity require additional detailed study.
Data Availability
The datasets generated during the current study are available from the corresponding author on reasonable request.
References
Austermann, J., J. Roth, and K. Barczyk-Kahlert. 2022. The good and the bad: Monocytes’ and macrophages’ diverse functions in inflammation. Cells 11: 1979. https://doi.org/10.3390/cells11121979. (PMID: 35741108).
Biswas, S.K., M. Chittezhath, I.N. Shalova, and J.Y. Lim. 2012. Macrophage polarization and plasticity in health and disease. Immunologic Research 53: 11–24. https://doi.org/10.1007/s12026-012-8291-9.
Blagov, A.V., A.M. Markin, A.I. Bogatyreva, T.V. Tolstik, V.N. Sukhorukov, and A.N. Orekhov. 2023. The role of macrophages in the pathogenesis of atherosclerosis. Cells 12 (4): 522. https://doi.org/10.3390/cells12040522.
Borges Da Silva, H., R. Fonseca, R.M. Pereira, A.A. Cassado, J.M. Álvarez, and M.R. D’Império Lima. 2015. Splenic macrophage subsets and their function during blood-borne infections. Frontiers in immunology 6: 480. https://doi.org/10.3389/fimmu.2015.00480. (PMID: 26441984).
Brown, I., M.G. Cascio, K.W.J. Wahle, R. Smoum, R. Mechoulam, R.A. Ross, R.G Pertwee and S.D. Heys. 2010. Cannabinoid receptor-dependent and -independent anti-proliferative effects of omega-3 ethanolamides in androgen receptor-positive and -negative prostate cancer cell lines. Carcinogenesis 31(9): 1584–1591. https://doi.org/10.1093/carcin/bgq151.
Calder, P.C. 2013. Omega-3 polyunsaturated fatty acids and inflammatory processes: Nutrition or pharmacology? British Journal of Clinical Pharmacology 75: 645–662. https://doi.org/10.1111/j.1365-2125.2012.04374.x. (PMID: 22765297).
Calvo, M.J., M.S. Martinez, W. Torres, M. Chavez-Castillo, E. Luzardo, N. Villasmil, J. Salazar, M. Velasco, and V. Bermudez. 2017. Omega-3 polyunsaturated fatty acids and cardiovascular health: a molecular view into structure and function. Vessel Plus 1: 116- 128. https://doi.org/10.20517/2574-1209.2017.14.
Cassetta, L., E. Cassol, and G. Poli. 2011. Macrophage polarization in health and disease. The Scientific World Journal 11: 2391–2402. https://doi.org/10.1100/2011/213962. (PMID: 22194670).
Chávez-Galán, L., M.L. Olleros, D. Vesin, and I. Garcia. 2015. Much more than M1 and M2 macrophages, there are also CD169+ and TCR+ macrophages. Frontiers in Immunology 6: 263. https://doi.org/10.3389/fimmu.2015.00263. (PMID: 26074923).
Chen, L., H. Deng, H. Cui, J. Fang, Z. Zuo, J. Deng, Y. Li, X. Wang, and L. Zhao. 2018. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 9: 7204–7218. https://doi.org/10.18632/oncotarget.23208. (PMID: 29467962).
Chen, S., A.F. Saeed, Q. Liu, Q. Jiang, H. Xu, G. Guishan Xiao, and L. Rao. 2023. Macrophages in immunoregulation and therapeutics. Signal Transduction and Targeted Therapy 8: 207. https://doi.org/10.1038/s41392-023-01452-1. (PMID: 37211559).
Cutolo, M., R. Campitiello, E. Gotelli, and S. Soldano. 2022. The role of M1/M2 macrophage polarization in rheumatoid arthritis synovitis. Frontiers in Immunology 13: 867260. https://doi.org/10.3389/fimmu.2022.867260. (PMID: 35663975).
Dzhalilova, D.S., A.M. Kosyreva, M.E. Diatroptov, N.A. Zolotova, I.S. Tsvetkov, V.A. Mkhitarov, O.V. Makarova, and D.N. Khochanskiy. 2019. Morphological characteristics of the thymus and spleen and the subpopulation composition of lymphocytes in peripheral blood during systemic inflammatory response in male rats with different resistance to hypoxia. International Journal of Inflammation 2019, 7584685. https://doi.org/10.1155/2019/7584685. (PMID: 31057785).
Deng, Z. and S. Liy. 2021. Inflammation-responsive delivery systems for the treatment of chronic inflammatory diseases. Drug Delivery and Translational Research 11: 1475–1497. https://doi.org/10.1007/s13346-021-00977-8. (PMID:33860447).
Fujiwara, N. and K. Kobayashi. Macrophages in inflammation. Current Drug Targets – Inflammation & Allergy 4: 281–286. https://doi.org/10.2174/1568010054022024. (PMID:16101534).
Garzetti L., R. Menon, A. Finardi, A. Bergami, A. Sica, G. Martino, G. Comi, C.Verderio, C. Farina, and R. Furlan. 2014. Activated macrophages release microvesicles containing polarized M1 or M2 mRNAs. Journal of Leukocyte Biology 95: 817–825. https://doi.org/10.1189/jlb.0913485. (PMID: 24379213).
Gentek, R., K. Molawi, and M.H. Sieweke. 2014. Tissue macrophage identity and self-renewal. Immunological Reviews, 262: 56–73. https://doi.org/10.1111/imr.12224. (PMID: 25319327).
Gordon, S. and P.R. Taylor. 2005. Monocyte and macrophage heterogeneity. Nature Reviews Immunology 5: 953–964. https://doi.org/10.1038/nri1733. (PMID: 16322748).
Han, X., S. Ding, H. Jiang, and G. Liu, 2021. Roles of macrophages in the development and treatment of gut inflammation. Frontiers in Cell and Developmental Biology 9: 625423. https://doi.org/10.3389/fcell.2021.625423. (PMID: 33738283).
Healy, D.A., F.A. Wallace, E.A. Miles, and P.C. Calder. 2000. The effect of low to moderate amounts of dietary fish oil on neutrophil lipid composition and function. Lipids 35: 763–768. https://doi.org/10.1007/s11745-000-0583-1. (PMID: 10941877).
Herold, K. and R. Mrowka. 2019. Inflammation-dysregulated inflammatory response and strategies for treatment. Acta Physiologica 226: Article e13284. https://doi.org/10.1111/apha.13284. (PMID: 31009174).
Hinz, B., S.H. Phan, V.J. Thannickal, M. Prunotto, A. Desmoulière, J. Varga, O. De Wever, M. Mareel, and G. Gabbiani. 2012. Recent developments in myofibroblast biology: Paradigms for connective tissue remodeling. The American Journal of Pathology 180: 1340–1355. https://doi.org/10.1016/j.ajpath.2012.02.004. (PMID: 22387320).
Jetten, N., S. Verbruggen, M.J. Gijbels, M.J. Post, M.P. De Winther, and M.M. Donners. 2014. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis 17: 109–118. https://doi.org/10.1007/s10456-013-9381-6. (PMID: 24013945).
Kim, H.Y., and A.A. Spector. 2018. N-Docosahexaenoylethanolamine: A neurotrophic and neuroprotective metabolite of docosahexaenoic acid. Molecular Aspects of Medicine 64: 34–44. https://doi.org/10.1016/j.mam.2018.03.004. (PMID: 29572109).
Kowal, K., R. Silver, E. Sławińska, M. Bielecki, L. Chyczewski, and O. Kowal-Bielecka. 2011. CD163 and its role in inflammation. Folia Histochemica et Cytobiologica 49: 365–374. https://doi.org/10.5603/fhc.2011.0052. (PMID: 22038213).
Krzyszczyk, P., R. Schloss, A. Palmer, and F. Berthiaume. 2018. The role of macrophages in acute and chronic wound healing and interventions to promote pro-wound healing phenotypes. Frontiers in Physiology 9: 419. https://doi.org/10.3389/fphys.2018.00419. (PMID: 29765329).
Latyshev, N.A., E.V. Ermolenko, and S.P. Kasyanov. 2014. Concentration and purification of polyunsaturated fatty acids from squid liver processing wastes. European Journal of Lipid Science and Technology 116: 1608–1613. https://doi.org/10.1002/ejlt.201400083.
Laria, A., A. Lurati, M. Marrazza, D. Mazzocchi, K.A. Re, and M. Scarpellin. 2016. The macrophages in rheumatic diseases. Journal of Inflammation Research 9: 1–11. https://doi.org/10.2147/JIR.S82320. (PMID: 26929657).
Lee, C.H., and E.Y. Choi. 2018. Macrophages and inflammation. Journal of Rheumatic Diseases 25: 11–18. https://doi.org/10.4078/jrd.2018.25.1.11.
Li, H., Y. Meng, S. He, X. Tan, Y. Zhang, X. Zhang, L. Wang, and W. Zheng. 2022. Macrophages, chronic inflammation, and insulin resistance. Cells 11: 3001. https://doi.org/10.3390/cells11193001. (PMID: 36230963).
Li, L., U. Maitra, N. Singh, and L. Gan. 2010. Molecular mechanism underlying LPS-induced generation of reactive oxygen species in macrophages. The FASEB Journal 24: 422.3–422.3. https://doi.org/10.1096/fasebj.24.1_supplement.422.3
Mantovani, A., A. Sica, and M. Locati. 2005. Macrophage polarization comes of age. Immunity 23: 344–346. https://doi.org/10.1016/j.immuni.2005.10.001. (PMID: 16226499).
Mantovani, A., S.K. Biswas, M.R. Galdiero, A. Sica, and M. Locati. 2013. Macrophage plasticity and polarization in tissue repair and remodeling. The Journal of Pathology 229: 176–185. https://doi.org/10.1002/path.4133c. (PMID: 23096265).
McKinney, M.K., and B.F. Cravatt. 2005. Structure and function of fatty acid amide hydrolase. Annual Review of Biochemistry 74: 411–432. https://doi.org/10.1146/annurev.biochem.74.082803.133450. (PMID: 15952893).
Medzhitov, R. 2010. Inflammation 2010: New adventures of an old flame. Cell 140: 771–776. https://doi.org/10.1016/j.cell.2010.03.006. (PMID: 20303867).
Meijerink, J., P. Plastina, J. Vincken, M. Poland, M. Attya, M. Balvers, H. Gruppen, B. Gabriele, and R.F. Witkamp. 2011. The ethanolamide metabolite of DHA, docosahexaenoylethanolamine, shows immunomodulating effects in mouse peritoneal and RAW264.7 macrophages: Evidence for a new link between fish oil and inflammation. British Journal of Nutrition 105: 1798–1807. https://doi.org/10.1017/S0007114510005635. (PMID: 21294934).
Meijerink, J., M. Balvers, and R. Witkamp. 2013. N-acyl amines of docosahexaenoic acid and other n–3 polyunsatured fatty acids – from fishy endocannabinoids to potential leads. British Journal of Pharmacology 169 (4): 772–783. https://doi.org/10.1111/bph.12030.
Meijerink, J., M. Poland, M.G.J. Balvers, P. Plastina, C. Lute, J. Dwarkasing, K. Norren, and R.F. Witkamp. 2014. Inhibition of COX-2-mediated eicosanoid production plays a major role in the anti-inflammatory effects of the endocannabinoid N-docosahexaenoylethanolamine (DHEA) in macrophages. British Journal of Pharmacology 172: 24–37. https://doi.org/10.1111/bph.12747.
Nishi, K., Y. Kanayama, I.H. Kim, A. Nakata, H. Nishiwaki, and T. Sugahara. 2019. Docosahexaenoyl ethanolamide mitigates IgE-mediated allergic reactions by inhibiting mast cell degranulation and regulating allergy-related immune cells. Scientific Reports 9: 16213. https://doi.org/10.1038/s41598-019-52317-z.
Park, T., H. Chen, K. Kevala, J.W. Lee, and H.Y. Kim. 2016. N-Docosahexaenoylethanolamine ameliorates LPS-induced neuroinflammation via cAMP/PKA-dependent signaling. Journal of Neuroinflammation 13: 284. https://doi.org/10.1186/s12974-016-0751-z. (PMID: 27809877).
Paterniti, I., D. Impellizzeri, R. Di Paola, E. Esposito, S. Gladman, P. Yip, J.V. Priestley, A.T. Michael-Titus, and S. Cuzzocrea. 2014. Docosahexaenoic acid attenuates the early inflammatory response following spinal cord injury in mice: In-vivo and in-vitro studies. Journal of Neuroinflammation 11: 6. https://doi.org/10.1186/1742-2094-11-6. (PMID: 24405628).
Porta, C., E. Riboldi, A. Ippolito, and A. Sica. 2015. Molecular and epigenetic basis of macrophage polarized activation. Seminars in Immunology 27: 237–248. https://doi.org/10.1016/j.smim.2015.10.003. (PMID: 26561250).
Romero-Sandoval, A., N. Nutile-McMenemy, and J.A. DeLeo. 2008. Spinal microglial and perivascular cell cannabinoid receptor type 2 activation reduces behavioral hypersensitivity without tolerance after peripheral nerve injury. Anesthesiology 108: 722–734. https://doi.org/10.1097/ALN.0b013e318167af74. (PMID: 18362605).
Rossol, M., H. Heine, U. Meusch, D. Quandt, C. Klein, M.J. Sweet, and S. Hauschildt. 2011. LPS-induced cytokine production in human monocytes and macrophages. Critical Reviews in Immunology 31: 379–446. https://doi.org/10.1615/critrevimmunol.v31.i5.20. (PMID: 22142165).
Rovito, D., C. Giordano, D. Vizza, P. Plastina, I. Barone, I. Casaburi, M. Lanzino, F. Amicis, D. Sisci, L. Mauro, S. Aquila, S. Catalano, D. Bonofiglio, and S. Andò. 2013. Omega-3 PUFA ethanolamides DHEA and EPEA induce autophagy through PPARg activation in MCF-7 breast cancer cells. Journal of Cellular Physiology 228 (6): 1314–1322. https://doi.org/10.1002/jcp.24288. (PMID: 23168911).
Saqib, U., S. Sarkar, K. Suk, O. Mohammad, M.S. Baig, and R. Savai. 2018. Phytochemicals as modulators of M1-M2 macrophages in inflammation. Oncotarget 9: 25; 17937–17950. https://doi.org/10.18632/oncotarget.24788. (PMID: 29707159).
Simon, F., and R. Fernández. 2009. Early lipopolysaccharide-induced reactive oxygen species production evokes necrotic cell death in human umbilical vein endothelial cells. Journal of Hypertension 27: 1202–1216. https://doi.org/10.1097/HJH.0b013e328329e31c. (PMID: 19307985).
Stulnig, T.M., and M. Zeyda. 2004. Immunomodulation by polyunsaturated fatty acids: Impact on T-cell signaling. Lipids 39: 1171–1175. https://doi.org/10.1007/s11745-004-1344-x. (PMID: 15736912).
Sung, J., H. Jeon, I. Kim, H.S. Jeong, and J. Lee. 2017. Anti-inflammatory effects of stearidonic acid mediated by suppression of NF-κB and MAP-kinase pathways in macrophages. Lipids 52: 781–787. https://doi.org/10.1007/s11745-017-4278-6. (PMID: 28744771).
Tang, R., Y.M. Lin, H.X. Liu, and E.S. Wang. 2018. Neuroprotective effect of docosahexaenoic acid in rat traumatic brain injury model via regulation of TLR4/NF-Kappa B signaling pathway. International Journal of Biochemistry and Cell Biology 99: 64–71. https://doi.org/10.1016/j.biocel.2018.03.017. (PMID: 29597004).
Tyrtyshnaia, A., S. Konovalova, A. Bondar, E. Ermolenko, R. Sultanov, and I. Manzhulo. 2021. Anti-inflammatory activity of N-docosahexaenoylethanolamine and N-eicosapentaenoylethanolamine in a mouse model of lipopolysaccharide-induced neuroinflammation. International Journal of Molecular Sciences 22: 10728. https://doi.org/10.3390/ijms221910728.
Tyrtyshnaia, A., S. Konovalova, A. Ponomarenko, A. Egoraeva, and I. Manzhulo. 2022. Fatty acid-derived N-acylethanolamines dietary supplementation attenuates neuroinflammation and cognitive impairment in LPS murine model. Nutrients 14: 3879. https://doi.org/10.3390/nu14183879. (PMID: 36145255).
Ueda, N., K. Tsuboi, and T. Uyama. 2010. N-acylethanolamine metabolism with special reference to N-acylethanolamine-hydrolysing acid amidase (NAAA). Progress in Lipid Research 49: 299–315. https://doi.org/10.1016/j.plipres.2010.02.003. (PMID: 20152858).
Yang, X., Y. Chang, and W. Wei. 2020. Emerging role of targeting macrophages in rheumatoid arthritis: focus on polarization, metabolism and apoptosis. Cell Proliferation 53: (7); Article e12854. https://doi.org/10.1111/cpr.12854. (PMID: 32530555).
Zeidler, P.C., L.M. Millecchia, and V. Castranova. 2004. Role of inducible nitric oxide synthase-derived nitric oxide in lipopolysaccharide plus interferon-gamma-induced pulmonary inflammation. Toxicology and Applied Pharmacology 195 (1): 45–54. https://doi.org/10.1016/j.taap.2003.10.005.
Zhou, D., C. Huang, Z. Lin, S. Zhan, L. Kong, C. Fang, and J. Li. 2014. Macrophage polarization and function with emphasis on the evolving roles of coordinated regulation of cellular signaling pathways. Cellular Signalling 26 (2): 192–197. https://doi.org/10.1016/j.cellsig.2013.11.004.
Funding
This research was funded by the Ministry of Science and Higher Education, Russian Federation (grant 13.1902.21.0012; contract no. 075–15-2020–796).
Author information
Authors and Affiliations
Contributions
Conceptualization: A.E., A.T., and I.M.; methodology: A.T., A.E., I.M., A.P., R.S., and D.I.; validation: A.E. and I.M.; formal analysis: A.E., A.T., and I.M.; investigation: A.T., A.E., I.M., A.P., R.S., and D.I.; resources: A.T., I.M., and R.S.; data curation: I.M.; writing—original draft preparation: A.E., I.M., and A.P.; writing—review and editing: A.E., A.T., and I.M.; visualization: I.M.; supervision: A.T. and I.M.; project administration: A.E. and I.M.; funding acquisition: I.M. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics Approval
All experimental procedures were approved by the Animal Ethics Committee at the A.V. Zhirmunsky National Scientific Center of Marine Biology, Far Eastern Branch, Russian Academy of Sciences (no. 1.3/2022) according to the Laboratory Animal Welfare guidelines and the European Communities Council Directive 2010/63/EU.
Informed Consent
Not applicable.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Egoraeva, A., Tyrtyshnaia, A., Ponomarenko, A. et al. Anti-inflammatory Effect of Polyunsaturated Fatty Acid N-Acylethanolamines Mediated by Macrophage Activity In Vitro and In Vivo. Inflammation 46, 2306–2319 (2023). https://doi.org/10.1007/s10753-023-01879-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-023-01879-2