
Abstract
Sepsis is a life-threatening condition. Polydatin (PD), a small natural compound from Polygonum cuspidatum, possesses antioxidant and anti-inflammatory properties. However, the protective mechanism of PD on sepsis-induced acute myocardial damage is still unclear. The aim of this study was to investigate the effect and mechanism of action of PD on lipopolysaccharide (LPS)-induced H9c2 cells and in a rat model of sepsis, and explored the role of PD-upregulated sirtuin (SIRT)6. LPS-induced H9c2 cells were used to simulate sepsis. Cecal ligation and puncture (CLP)-induced sepsis in rats were used to verify the protective effect of PD. ELISA, western blotting, immunofluorescence, immunohistochemistry, and flow cytometry were used to study the protective mechanism of PD against septic myocardial injury. PD pretreatment suppressed LPS-induced H9c2 cell apoptosis by promotion of SIRT6-mediated autophagy. Downregulation of SIRT6 or inhibition of autophagy reversed the protective effect of PD on LPS-induced apoptosis. PD pretreatment also suppressed LPS-induced inflammatory factor expression. CLP-induced sepsis in rats showed that PD pretreatment decreased CLP-induced myocardial apoptosis and serum tumor necrosis factor-α, interleukin (IL)-1β, and IL-6 expression. 3-Methyladenine (autophagy inhibitor) pretreatment prevented the protective effect of PD on septic cardiomyopathy. SIRT6 expression was increased with PD treatment, which confirmed that PD attenuates septic cardiomyopathy by promotion of SIRT6-mediated autophagy. All these results indicate that PD has potential therapeutic effects that alleviate septic myocardial injury by promotion of SIRT6-mediated autophagy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Severe sepsis and septic shock are major healthcare problems throughout the world annually [1]. Multiple organ failure in sepsis is a consequence of cellular dysfunction in the affected organs [2, 3], and there is compelling evidence that cardiovascular dysfunction is a major complication associated with sepsis-induced morbidity and mortality [4]. Polydatin (PD), a glucoside of resveratrol, known as a monocrystalline compound isolated from Polygonum cuspidatum Sieb. et Zucc., is used for both medication and food [5]. Increasing evidence shows that PD has strong anti-inflammatory and antioxidative bioactivity [6,7,8]. PD inhibits mitochondrial dysfunction in the renal tubular epithelial cells of a rat model of sepsis-induced acute kidney injury [9]. PD also has protective effects on septic lung injury in mice via upregulation of heme oxygenase-1 [10]. Whether PD has a protective effect on sepsis-induced acute myocardial damage is still unclear.
Autophagy is a cellular degradation process that is distinct from the ubiquitin–proteasome system, and is a primary mechanism for maintaining cellular homeostasis. Increasing evidence shows a relationship between autophagy and apoptosis [11,12,13]. Some studies have found that autophagy plays an important role in cell survival after sepsis [14, 15]. Previous studies have been found that valproic acid can attenuate sepsis-induced myocardial dysfunction in rats by accelerating autophagy through the PTEN/AKT/mTOR pathway [16]. MiR-155 alleviates septic lung injury by inducing autophagy via inhibition of transforming growth factor-β-activated binding protein 2 [17]. However, excessive autophagy can cause unwanted and deleterious cell death [18]. PD regulates cell proliferation, apoptosis, and autophagy [19], but the specific mechanism is not clear.
Sirtuin (SIRT)6, which belongs to the sirtuin family of NAD+-dependent enzymes, has many pivotal functions and exhibits multiple enzymatic activities [20]. Expression of SIRT6 promotes activation of autophagy [21, 22]. Previous studies have found that sirt6-induced autophagy restricted TREM-1-mediated pyroptosis in ox-LDL-treated endothelial cells: relevance to prognostication of patients with acute myocardial infarction [23]. PD ameliorates diabetic cardiomyopathy via SIRT3 activation [24]. Both SIRT3 and SIRT6 belong to the NAD+-dependent class III deacetylase sirtuin family [25]. It is suggested that PD alleviates septic myocardial injury by promoting SIRT6-mediated autophagy.
In the present study, we investigated the function and potential mechanism of action of PD on sepsis-mediated cardiomyopathy and elucidated the protective mechanism of PD through a cecal ligation and puncture (CLP) rat model and lipopolysaccharide (LPS)-induced H9c2 cell damage.
MATERIALS AND METHODS
Ethics Statement
All animal experiments were approved by the Animal Ethics Committee of Pudong New Area Gongli Hospital (Shanghai, China).
Sepsis Model and Treatment
Male Sprague-Dawley rats (age 7–8 weeks and weighing 200–250 g) were provided by Shanghai SLAC Laboratory Animal Co. (Shanghai, China). Rats were provided with irradiated food, free access to sterile acidified water, and were housed in individual microisolators. For sepsis induction, animals were anesthetized by intraperitoneal injection of 2% sodium pentobarbital in saline (40 mg/kg; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) and placed on a warming pad (37 °C). CLP was performed as previous studies [9]. The rat abdomen was shaved before a 2-cm midline abdominal incision was made. To prevent bowel obstruction, the cecum was carefully isolated, and ~ 60% of the total cecum length was ligated below the ileocecal valve. Two punctures were made on the opposite side of the mesenteric with a sterile 18-gauge needle. The cecum was gently pressed to expel a small amount of stool from the puncture site to ensure a full-thickness perforation. The cecum was returned to the peritoneal cavity, and the abdominal incision was closed in two layers. The ceca of sham-operated rats were not ligated or punctured, but underwent the same aforementioned procedure. All rats were subcutaneously injected with normal saline (5 ml/100 g) immediately following surgery in order to resuscitate. They were then group-housed in a temperature-controlled room (22 °C) in cages with dry sawdust bedding. Fluid blocks and soft food were provided. The mortality and behavioral signs were monitored and recorded every day following the CLP procedure.
To investigate the effect of PD and autophagy on sepsis-induced cardiomyopathy, PD (30 mg/kg, Sigma-Aldrich, St. Louis, MO, USA) [9] or the autophagic inhibitor, 3-methyladenine (3-Ma, 20 mg/kg; Sigma-Aldrich) was administered intraperitoneally 6 h before CLP. An equal dose of normal saline was administered to the control group. Each group comprised ten rats. At 48 h after surgery, the hearts were collected for subsequent experiments after statistical analysis of survival.
Echocardiographic and Hemodynamic Measurements
We anesthetized rats with intramuscular injections of ketamine (100 mg/kg) and medetomidine (0.25 mg/kg). Cardiac function parameters such as left ventricular internal dimension in systole (LVID(s)), left ventricular internal dimension in diastole (LVID(d)), left ventricular fraction shortening (LVFS), and left ventricular ejection fraction (LVEF) were captured by transthoracic echocardiography system, which had a 15-MHz phased-array transducer (SONOS 5500; Hewlett-Packard, Andover, MA, USA). We inserted a catheter into the rat left ventricle from the right carotid artery. In this manner, the left ventricular end diastolic pressure (LVEDP), heart rate (HR), and cardiac contractility (dP/dt maximum and dP/dt minimum) were assayed and recorded with a PowerLab ML880 (AD Instruments, Bella Vista, NSW, Australia).
Cell Lines and Cell Culture
The cardiomyoblast cell line H9c2 was purchased from the American Type Culture Collection (Manassas, VA, USA) and cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. To investigate whether SIRT6 plays a role in sepsis-mediated myocardial protection and to clarify its mechanism of action, H9c2 cells were transfected with siRNA against SIRT6 (constructed by Genechem, Shanghai, China) for 48 h before induction with LPS (1 μg/ml) for 24 h. H9c2 cells were harvested for biological analysis. To investigate the role of autophagy in sepsis-mediated myocardial damage, H9c2 cells transfected with GFP-LC3 vector (Genechem, Shanghai, China) were pretreated with PD (100 μM) or 3-Ma (10 μM) for 30 min before induction with LPS (1 μg/ml) for 24 h.
Immunofluorescence and Immunohistochemistry
The myocardial sections and H9c2 cells were deparaffinized and heated in citrate buffer for antigen retrieval. Phosphate-buffered saline containing 10% fetal bovine serum was added to each section to reduce background staining for 30 min at room temperature. Sections of 5 μm were used for terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining for apoptosis analysis with an Axiophot light microscope (Zeiss, Oberkochen, Germany). For autophagy analysis, H9c2 cells were stained with LC3 antibody and analyzed by fluorescence microscopy (Nikon, Tokyo, Japan) and photographed with a digital camera.
Flow Cytometry
Flow cytometry was used to determine the rate of apoptosis of H9c2 cells. Apoptotic cells were differentiated from viable or necrotic cells by the combined application of annexin V (AV)–fluorescein isothiocyanate (FITC) and propidium iodide (PI). Cells were washed twice and adjusted to a concentration of 106 cells/ml with cold D-Hanks buffer. AV–FITC (10 μl) and PI (10 μl) were added to 100 μl of cell suspension and incubated for 15 min at room temperature in the dark. Finally, 400 μl of binding buffer was added to each sample without washing and analyzed using flow cytometry. Each experiment was performed at least in three independent experiments.
Western Blot Analysis
Cells or tissues were lysed; protease inhibitors were added to the lysates; and cell lysate was centrifuged at 12000 rpm at 4 °C. The protein concentration was examined by the BCA kit (Pierce, Rockford, IL, USA). The protein was separated using 10% SDS-PAGE assay and moved to a PVDF membrane. Antibodies listed below were applied to determine the protein expression: SIRT6 (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA, USA), cleaved caspase-3 (1:1000; Santa Cruz Biotechnology), caspase-3 (1:1000; Santa Cruz Biotechnology), Beclin-1 (1:500; Santa Cruz Biotechnology), P62 (1:500; Santa Cruz Biotechnology), LC3 (1:500; Santa Cruz Biotechnology), Bcl-2 (1:500; Santa Cruz Biotechnology), Bax (1:1000; Santa Cruz Biotechnology), GAPDH (1:1000; Santa Cruz Biotechnology), and horseradish-peroxidase-conjugated secondary antibody (1:1000; Abcam, Cambridge, MA, USA). An ECL chemiluminescent kit (Millipore, Billerica, MA, USA) was applied to measure the bands.
ELISA for Soluble Inflammatory Cytokines
The amounts of interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF)-α in the supernatants of H9c2 cells or serum from rats were measured using commercially available ELISA kits (Sen-Xiong Company). Supernatants were stored at − 80 °C before measurement, and both standards and samples were run in triplicate. OD450 was calculated by subtracting the background, and standard curves were plotted.
Statistical Analysis
All data are expressed as the mean ± SEM. Data analysis was performed using GraphPad Prism 5.0 (GraphPad, La Jolla, CA, USA). When analyzing two groups, Student’s t test was used to determine statistical significance. When comparing multiple groups, one-way analysis of variance was used, followed by Tukey’s multiple comparisons test to determine statistically significant differences. P < 0.05 was considered significant.
RESULTS
PD Pretreatment Attenuated LPS-Induced Inflammation and Apoptosis of H9c2 Cells by Promotion of Autophagy
PD is a monocrystalline and polyphenolic drug isolated from a traditional Chinese herb (P. cuspidatum). It is protective against cell dysfunction induced by inflammatory factors and has been approved for clinical trials for treatment of diabetic cardiomyopathy [24] and myocardial infarction [26, 27]. The mechanism of the protective effects of PD on sepsis-induced acute myocardial damage is still unclear. H9c2 cells were used to study the protective effect of PD on LPS-induced myocardial cell damage. H9c2 cell apoptosis was increased after simulation of sepsis conditions with LPS (1 μg/ml) for 24 h. PD pretreatment decreased LPS-induced apoptosis. Inhibition of autophagy with 3-Ma pretreatment reversed the protective effect of PD on LPS-induced H9c2 cell apoptosis (Fig. 1a, b). Western blotting showed that PD pretreatment downregulated LPS-induced caspase-3 and Bax expression but promoted Bcl-2 expression. 3-Ma pretreatment suppressed PD-induced Bcl-2 expression and promoted cleaved caspase-3 and Bax expression (Fig. 1c–f). ELISA showed that PD pretreatment decreased LPS-induced IL-1β, IL-6, and TNF-α expression. The inhibitory effects of PD were reversed by pretreatment with 3-Ma (Fig. 1g–i). It is suggested that autophagy is involved in the PD-mediated protective effect against LPS-induced H9c2 cell damage, which was confirmed by immunofluorescence. PD pretreatment promoted LPS-induced autophagy plaque formation (Fig. 1j, k). Western blotting showed that PD pretreatment promoted LPS-induced, autophagy-related protein LC3-II and Beclin-1 expression, but decreased P62 expression (Fig. 1l–o).

PD treatment attenuated LPS-induced inflammation and apoptosis of H9c2 cells by promotion of autophagy. H9c2 cells were treated with LPS (1 μg/ml) after pretreatment with PD (100 μM) combined with or without autophagy inhibitor 3-MA (10 μM) for 24 h. a Apoptosis was analyzed by flow cytometry after double labeling with AV–FITC and PI. b the percentage of apoptotic cells in each group was analyzed. Data are presented as the mean ± SD. ***P < 0.001 versus normal control. ###P < 0.001 versus LPS treatment group. c–f Western blotting showed expression of cleaved caspase-3, caspase-3, Bcl-2, and Bax. GAPDH was used as an endogenous control. Data are presented as the mean ± SD. ***P < 0.001 versus the normal controls. ###P < 0.001 versus LPS treatment group. g–i inflammatory factors IL-1β, IL-6, and TNF-α from the supernatant were detected by ELISA. Data are presented as the mean ± SD. ***P < 0.001 versus normal controls. ###P < 0.001 versus LPS treatment group. j and k Immunofluorescence showed autophagy puncta in H9c2 cells. Scale bar, 20 μm. Data presented as mean ± SD. ***P < 0.001 versus controls. ###P < 0.001 versus LPS treatment group. l–o Western blotting showed expression of P62, LC3, and Beclin-1. Data presented as mean ± SD. ***P < 0.001 versus normal controls. ###P < 0.001 versus LPS treatment group.
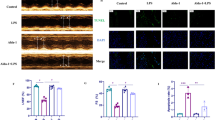
Downregulation of SIRT6 Reversed the Suppressive Effect of PD on LPS-Induced Inflammation and Apoptosis of H9c2 Cells by Inhibition of Autophagy
Increasing evidence shows that SIRT6 overexpression has a protective effect against stress-induced apoptosis [22]. To confirm whether SIRT6 was involved in PD-mediated protection against LPS-induced H9c2 cell damage, siRNA against SIRT6 was constructed. RT-PCR and western blotting showed that SIRT6 expression was downregulation after SIRT6 silencing when compared with control or negative control groups (Fig. 2a, b). Apoptosis was analyzed by flow cytometry, which showed that downregulation SIRT6 suppressed the protective effect of PD on survival of H9c2 cells treated with LPS condition (Fig. 2c, d). Western blotting confirmed that silencing SIRT6 decreased the inhibitory effect of PD on cleaved caspase-3 and Bax expression. Downregulation of SIRT6 expression also decreased Bcl-2 expression, even with PD pretreatment (Fig. 2e–h). ELISA showed that downregulation of SIRT6 expression reversed PD-induced inhibition of inflammatory factor IL-1β, IL-6, and TNF-α expression under LPS treatment (Fig. 2i–k).

Downregulation of SIRT6 reversed the suppressive effect of PD on LPS-induced inflammation and apoptosis of H9c2 cells. a and b RT-PCR and western blotting showed expression of SIRT6 in H9c2 cells after transfection with siRNA against SIRT6 (siSIRT6) or negative control (NC) for 48 h. Data are presented as the mean ± SD. ***P < 0.001 versus controls. c Apoptosis was analyzed by flow cytometry after double labeling with AV–FITC and PI. d The percentage of apoptotic cells in each group was analyzed. Data are presented as the mean ± SD. ***P < 0.001 versus LPS group. ###P < 0.001 versus LPS + PD treatment group. e–h Western blotting showed expression of caspase-3, Bcl-2, and Bax. GAPDH was used as an endogenous control. Data are presented as the mean ± SD. ***P < 0.001 versus the controls. ###P < 0.001 versus LPS treatment group. i–k Inflammatory factors IL-1β, IL-6, and TNF-α from supernatant were detected by ELISA. Data are presented as the mean ± SD. ***P < 0.001 versus LPS group. ###P < 0.001 versus LPS + PD treatment group.
To confirm that SIRT6 had a regulatory effect on autophagy, immunofluorescence showed that PD pretreatment promoted autophagy under LPS treatment. Silencing of SIRT6 suppressed autophagy, even with PD pretreatment (Fig. 3a, b). Western blotting confirmed that downregulation of SIRT6 suppressed Beclin-1 expression and reduced the ratio of LC3-II/LC3-I. Silencing SIRT6 promoted P62 expression, which had inhibitory effect on autophagy (Fig. 3c–f).

SIRT6 played an important role in PD-induced autophagy under LPS treatment. a and b Immunofluorescence showed autophagy puncta in H9c2 cells. Data presented as mean ± SD. Scale bar, 20 μm. ***P < 0.001 versus LPS group. ###P < 0.001 versus LPS + PD treatment group. c–f Western blotting showed expression of P62, LC3, and Beclin-1. Data presented as mean ± SD. ***P < 0.001 versus LPS group. ###P < 0.001 versus LPS + PD treatment group.
PD Pretreatment Attenuated Sepsis-Induced Myocardial Apoptosis and Ameliorated Survival in Rats
To establish whether PD had a protective effect against sepsis, PD (30 mg/kg) or 3-Ma (20 mg/kg) was administered intraperitoneally 6 h before CLP. Each group had 6 rats. After CLP, echocardiography showed significantly decreased LVFS and LVEF, while LVID significantly increased compared with sham rats; importantly, these changes were partially alleviated by PD treatment. However, after autophagy activation suppressed with 3-Ma treatment, the protective effect of PD was reversed. Hemodynamic examination demonstrated that CLP-mediated sepsis decreased dP/dt and increased LVEDP, which were both ameliorated by PD treatment; however, after autophagy activation suppressed with 3-Ma treatment, the protective effect of PD was reversed (Table 1).
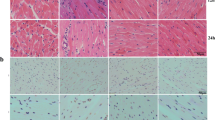
Immunohistochemical staining showed that myocardial apoptosis was increased after CLP for 48 h when compared with normal controls. After PD pretreatment, myocardial apoptosis was decreased compared with the CLP group. 3-Ma pretreatment decreased the protective effect of PD against CLP-induced myocardial damage (Fig. 4a, b). PD pretreatment decreased sepsis-induced myocardial apoptosis by promotion of autophagy. Western blotting confirmed that PD pretreatment suppressed sepsis-induced cleaved caspase-3 and Bax expression in myocardial tissue. PD pretreatment promoted Bcl-2 expression (Fig. 4c–f).

PD attenuated sepsis-induced myocardial apoptosis and ameliorated survival in rats. a and b Myocardial apoptosis was measured using TUNEL staining of heart tissue sections. Data are represented as mean ± SD. ***P < 0.001 versus control group. ###P < 0.001 versus CLP group. c–f Western blotting showed expression of caspase-3, Bax, and Bcl-2. GAPDH was used as an endogenous control. Data are presented as mean ± SD. **P < 0.01, ***P < 0.001 versus control group. ###P < 0.001 versus CLP group. e The survival of rats was calculated after CLP and 0, 6, 12, 24, 36, and 48 h pretreatment with or without PD (30 mg/kg) or 3-Ma (20 mg/kg).
Mortality was monitored from 4 to 48 h before the rats were killed. Mortality of rats with CLP-induced sepsis was increased. PD pretreatment suppressed death after CLP. Pretreatment with 3-Ma decreased the protective effect of PD against CLP-induced death of rats in the acute phase of sepsis (Fig. 4g).
PD Pretreatment Decreased Serum Inflammatory Factor Levels in Rats Subjected to CLP by Promotion of SIRT6 Activation and Myocardial Autophagy
Serum inflammatory factors IL-1β, IL-6, and TNF-α were detected by ELISA after CLP for 48 h. PD pretreatment decreased CLP-induced serum inflammatory factor levels. 3-Ma pretreatment alleviated the inhibitory effect of PD on CLP-induced inflammatory factor expression (Fig. 5). Western blotting showed that expression of SIRT6 was downregulated after CLP. PD pretreatment promoted SIRT6 expression. 3-Ma pretreatment had no effect on PD-induced SIRT6 expression (Fig. 6a, b). It was suggested that SIRT6 had a regulatory effect on autophagy, but suppression of autophagy had no effect on SIRT6 expression. Western blotting confirmed that PD pretreatment promoted Beclin-1 expression and increased the ratio of LC3-II/LC3-I. PD pretreatment decreased P62 expression (Fig. 6c–f).

PD decreased serum inflammatory factor levels in rats subjected to CLP. a–c Serum inflammatory factors IL-6 (a), IL-1β (b), and TNF-α (c) were detected by ELISA. Data are presented as the mean ± SD. **P < 0.01, ***P < 0.001 versus control group. ###P < 0.001 versus LPS treatment group.

PD promoted SIRT6 activation and myocardial autophagy. a and b Western blotting showed expression of SIRT6. GAPDH was used as an endogenous control. Data are presented as the mean ± SD. ***P < 0.001 versus control group. ###P < 0.001 versus CLP group. c–f Western blotting showed expression of P62, LC3, and Beclin-1. GAPDH was used as an endogenous control. Data are presented as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001 versus control group. #P < 0.05, #P < 0.01, ###P < 0.001 versus CLP group.
DISCUSSION
Sepsis-induced myocardial dysfunction (SIMD) is reversible myocardial depression caused by sepsis [28, 29]. Although several studies have reported that endotoxins, inflammatory cytokines, and nitric oxide are related to the pathogenesis of SIMD, the condition is still not completely understood [30]. In this study, we found that autophagy was involved in the progression of SIMD. In the acute stage, suppressed 3-Ma can aggravate the disease by promotion of apoptosis and inflammatory factor expression. This was consistent with previous reports that autophagy activation ameliorates sepsis-induced organ damage [31, 32]. PD pretreatment significantly decreased sepsis-induced myocardial apoptosis and inflammatory factor expression. PD also promotes autophagy activation. Inhibition of autophagy by 3-Ma reversed the protective effect of PD against SIMD.
We found that PD ameliorated SIMD by reducing myocardial apoptosis and inflammatory factor expression. Previous studies have found that PD prevents induction of secondary brain injury after traumatic brain injury by protecting neuronal mitochondria (Li et al., 2019). PD inhibits proliferation and promotes apoptosis of doxorubicin-resistant osteosarcoma through lncRNA TUG1-mediated suppression of Akt signaling [33]. In sepsis, PD inhibited mitochondrial dysfunction in the renal tubular epithelial cells of a rat model of sepsis-induced acute kidney injury [9]. PD also induces septic lung injury in mice via upregulation of HO-1 [10, 34]. In this study, we also found that SIRT6 was involved in autophagy regulation after PD treatment. During the past few years, SIRT6 has been investigated extensively, and current evidence shows that it participates in regulation of metabolism, inflammation, and apoptosis [35, 36]. We found that PD pretreatment promoted SIRT6 expression. Silencing SIRT6 significantly suppressed autophagy activation and decreased the protective effect of PD against SIMD. We suggest that PD alleviates septic myocardial injury, probably by promotion of SIRT6-mediated autophagy. Previous studies have found that Sirt6 protects podocytes from apoptosis and inflammation by increasing autophagic flux through inhibition of the Notch pathway [37]. Blockage of Sirt6-induced autophagy leads to augmented TREM-1-mediated pyroptosis [23]. However, the specific follow-up mechanism for SIRT6 in autophagy needs to be further studied.
In conclusion, our results demonstrate that PD suppresses SIMD via inhibition of myocardial apoptosis and inflammatory factor expression probably by promotion of SIRT6-mediated autophagy. PD might be used as a novel treatment for sepsis.
References
Mosevoll, K.A., S. Skrede, D.L. Markussen, H.R. Fanebust, H.K. Flaatten, J. Assmus, et al. 2018. Inflammatory mediator profiles differ in sepsis patients with and without bacteremia. Frontiers in Immunology 9: 691.
Marmanillo, C.G., C. Langaro, J.E. Nicoluzzi, R.T. Belila, M. Macri, R. Zamprogna, M. Luvizotto, and M. Takahashi. 2018. Renopancreatic transplantation: evaluation of 15 years in 131 patients. Transplantation Proceedings 50: 792–795.
Zhao, Y., H. Zhu, H. Wang, L. Ding, L. Xu, D. Chen, S. Shen, Y. Hou, and H. Dou. 2018. FC-99 ameliorates sepsis-induced liver dysfunction by modulating monocyte/macrophage differentiation via let-7a related monocytes apoptosis. Oncotarget 9: 14959–14976.
Ma, H., X. Wang, T. Ha, M. Gao, L. Liu, R. Wang, K. Yu, J.H. Kalbfleisch, R.L. Kao, D.L. Williams, and C. Li. 2016. MicroRNA-125b prevents cardiac dysfunction in polymicrobial sepsis by targeting TRAF6-mediated nuclear factor kappaB activation and p53-mediated apoptotic signaling. The Journal of Infectious Diseases 214: 1773–1783.
Huang, Q.H., L.Q. Xu, Y.H. Liu, J.Z. Wu, X. Wu, X.P. Lai, et al. 2017. Polydatin protects rat liver against ethanol-induced injury: involvement of CYP2E1/ROS/Nrf2 and TLR4/NF-kappaB p65 pathway. Evidence-based Complementary and Alternative Medicine 2017: 7953850.
Pang, N., T. Chen, X. Deng, N. Chen, R. Li, M. Ren, et al. 2017. Polydatin prevents methylglyoxal-induced apoptosis through reducing oxidative stress and improving mitochondrial function in human umbilical vein endothelial cells. Oxidative Medicine and Cellular Longevity 2017: 7180943.
Ye, J., H. Piao, J. Jiang, G. Jin, M. Zheng, J. Yang, et al. 2017. Polydatin inhibits mast cell-mediated allergic inflammation by targeting PI3K/Akt, MAPK, NF-kappaB and Nrf2/HO-1 pathways. Scientific Reports 7: 11895.
Tang, S., Q. Tang, J. Jin, G. Zheng, J. Xu, W. Huang, X. Li, P. Shang, and H. Liu. 2018. Polydatin inhibits the IL-1beta-induced inflammatory response in human osteoarthritic chondrocytes by activating the Nrf2 signaling pathway and ameliorates murine osteoarthritis. Food & Function 9: 1701–1712.
Gao, Y., Z. Zeng, T. Li, S. Xu, X. Wang, Z. Chen, and C. Lin. 2015. Polydatin inhibits mitochondrial dysfunction in the renal tubular epithelial cells of a rat model of sepsis-induced acute kidney injury. Anesthesia and Analgesia 121: 1251–1260.
Li, X.H., X. Gong, L. Zhang, R. Jiang, H.Z. Li, M.J. Wu, et al. 2013. Protective effects of polydatin on septic lung injury in mice via upregulation of HO-1. Mediators of Inflammation 2013: 354087.
Booth, L.A., S. Tavallai, H.A. Hamed, N. Cruickshanks, and P. Dent. 2014. The role of cell signalling in the crosstalk between autophagy and apoptosis. Cellular Signalling 26: 549–555.
Denton, D., T. Xu, and S. Kumar. 2015. Autophagy as a pro-death pathway. Immunology and Cell Biology 93: 35–42.
Gump, J.M., L. Staskiewicz, M.J. Morgan, A. Bamberg, D.W. Riches, and A. Thorburn. 2014. Autophagy variation within a cell population determines cell fate through selective degradation of Fap-1. Nature Cell Biology 16: 47–54.
Kader, M., M. Alaoui-El-Azher, J. Vorhauer, B.B. Kode, J.Z. Wells, D. Stolz, et al. 2017. MyD88-dependent inflammasome activation and autophagy inhibition contributes to Ehrlichia-induced liver injury and toxic shock. PLoS Pathogens 13: e1006644.
Sunahara, S., E. Watanabe, M. Hatano, P.E. Swanson, T. Oami, L. Fujimura, et al. 2018. Influence of autophagy on acute kidney injury in a murine cecal ligation and puncture sepsis model. Scientific Reports 8: 1050.
Shi, X., Y. Liu, D. Zhang, and D. Xiao. 2019. Valproic acid attenuates sepsis-induced myocardial dysfunction in rats by accelerating autophagy through the PTEN/AKT/mTOR pathway. Life Sciences 232: 116613.
Liu, F., C. Nie, N. Zhao, Y. Wang, Y. Liu, Y. Li, Z. Zeng, C. Ding, Q. Shao, C. Qing, L. Xia, Z. Peng, and K. Qian. 2017. MiR-155 alleviates septic lung injury by inducing autophagy via inhibition of transforming growth factor-beta-activated binding protein 2. Shock 48: 61–68.
Crowell, K.T., D.I. Soybel, and C.H. Lang. 2017. Inability to replete white adipose tissue during recovery phase of sepsis is associated with increased autophagy, apoptosis, and proteasome activity. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology 312: R388–R399.
Yang, B., and S. Zhao. 2017. Polydatin regulates proliferation, apoptosis and autophagy in multiple myeloma cells through mTOR/p70s6k pathway. OncoTargets and Therapy 10: 935–944.
Zhang, X., S. Khan, H. Jiang, M.A. Antonyak, X. Chen, N.A. Spiegelman, J.H. Shrimp, R.A. Cerione, and H. Lin. 2016. Identifying the functional contribution of the defatty-acylase activity of SIRT6. Nature Chemical Biology 12: 614–620.
Feng, Y., J. Liang, Y. Zhai, J. Sun, J. Wang, X. She, Q. Gu, Y. Liu, H. Zhu, X. Luo, and X. Sun. 2018. Autophagy activated by SIRT6 regulates Abeta induced inflammatory response in RPEs. Biochemical and Biophysical Research Communications 496: 1148–1154.
Chen, J., J.J. Xie, M.Y. Jin, Y.T. Gu, C.C. Wu, W.J. Guo, et al. 2018. Sirt6 overexpression suppresses senescence and apoptosis of nucleus pulposus cells by inducing autophagy in a model of intervertebral disc degeneration. Cell Death & Disease 9: 56.
Zi, Y., Y. Yi-An, J. Bing, L. Yan, T. Jing, G. Chun-Yu, et al. 2019. Sirt6-induced autophagy restricted TREM-1-mediated pyroptosis in ox-LDL-treated endothelial cells: relevance to prognostication of patients with acute myocardial infarction. Cell Death Discovery 5: 88.
Zhang, M., S. Wang, Z. Cheng, Z. Xiong, J. Lv, Z. Yang, T. Li, S. Jiang, J. Gu, D. Sun, and Y. Fan. 2017. Polydatin ameliorates diabetic cardiomyopathy via Sirt3 activation. Biochemical and Biophysical Research Communications 493: 1280–1287.
Tanabe, K., J. Liu, D. Kato, H. Kurumizaka, K. Yamatsugu, M. Kanai, et al. 2018. LC-MS/MS-based quantitative study of the acyl group- and site-selectivity of human sirtuins to acylated nucleosomes. Scientific Reports 8: 2656.
Zhang, M., Z. Zhao, M. Shen, Y. Zhang, J. Duan, Y. Guo, et al. 1863. Polydatin protects cardiomyocytes against myocardial infarction injury by activating Sirt3. Biochimica et Biophysica Acta - Molecular Basis of Disease 2017: 1962–1972.
Gao, Y., J. Gao, C. Chen, H. Wang, J. Guo, and R. Wu. 2015. Cardioprotective effect of polydatin on ventricular remodeling after myocardial infarction in coronary artery ligation rats. Planta Medica 81: 568–577.
Jeong, H.S., T.H. Lee, C.H. Bang, J.H. Kim, and S.J. Hong. 2018. Risk factors and outcomes of sepsis-induced myocardial dysfunction and stress-induced cardiomyopathy in sepsis or septic shock: a comparative retrospective study. Medicine (Baltimore) 97: e0263.
Vieillard-Baron, A., V. Caille, C. Charron, G. Belliard, B. Page, and F. Jardin. 2008. Actual incidence of global left ventricular hypokinesia in adult septic shock. Critical Care Medicine 36: 1701–1706.
Zaky, A., S. Deem, K. Bendjelid, and M.M. Treggiari. 2014. Characterization of cardiac dysfunction in sepsis: an ongoing challenge. Shock 41: 12–24.
Cao, C., T. Gao, Y. Cheng, M. Cheng, T. Su, F. Xi, C. Wu, and W. Yu. 2018. Hypothalamic AMPK-induced autophagy ameliorates hypercatabolism in septic rats by regulating POMC expression. Biochemical and Biophysical Research Communications 497: 1089–1096.
Inata, Y., S. Kikuchi, R.S. Samraj, P.W. Hake, M. O’Connor, J.R. Ledford, J. O’Connor, P. Lahni, V. Wolfe, G. Piraino, and B. Zingarelli. 2018. Autophagy and mitochondrial biogenesis impairment contribute to age-dependent liver injury in experimental sepsis: dysregulation of AMP-activated protein kinase pathway. The FASEB Journal 32: 728–741.
Hu, T., Z. Fei, H. Su, R. Xie, and L. Chen. 2019. Polydatin inhibits proliferation and promotes apoptosis of doxorubicin-resistant osteosarcoma through LncRNA TUG1 mediated suppression of Akt signaling. Toxicology and Applied Pharmacology 371: 55–62.
Li, L., H.P. Tan, C.Y. Liu, L.T. Yu, D.N. Wei, Z.C. Zhang, K. Lu, K.S. Zhao, M. Maegele, D.Z. Cai, and Z.T. Gu. 2019. Polydatin prevents the induction of secondary brain injury after traumatic brain injury by protecting neuronal mitochondria. Neural Regeneration Research 14: 1573–1582.
Han, Z., L. Liu, Y. Liu, and S. Li. 2014. Sirtuin SIRT6 suppresses cell proliferation through inhibition of Twist1 expression in non-small cell lung cancer. International Journal of Clinical and Experimental Pathology 7: 4774–4781.
Marquardt, J.U., K. Fischer, K. Baus, A. Kashyap, S. Ma, M. Krupp, M. Linke, A. Teufel, U. Zechner, D. Strand, S.S. Thorgeirsson, P.R. Galle, and S. Strand. 2013. Sirtuin-6-dependent genetic and epigenetic alterations are associated with poor clinical outcome in hepatocellular carcinoma patients. Hepatology 58: 1054–1064.
Liu, M., K. Liang, J. Zhen, M. Zhou, X. Wang, Z. Wang, et al. 2017. Sirt6 deficiency exacerbates podocyte injury and proteinuria through targeting notch signaling. Nature Communications 8: 413.
Funding
This study was supported by the Innovation Funds for Development of Science and Technology in PuDong New Area (PKJ2017-Y22) and the Supported projects for the construction of key and weak disciplines in Pudong New Area (PWZbr2017-08).
Author information
Authors and Affiliations
Contributions
Dongfeng Guo generated and analyzed the data; Xiaoyan Yuan and Guo Chen designed the experiments and drafted the manuscript. All authors approved the version of the manuscript to be published.
Corresponding authors
Ethics declarations
All animal experiments were approved by the Animal Ethics Committee of Pudong New Area Gongli Hospital (Shanghai, China).
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yuan, X., Chen, G., Guo, D. et al. Polydatin Alleviates Septic Myocardial Injury by Promoting SIRT6-Mediated Autophagy. Inflammation 43, 785–795 (2020). https://doi.org/10.1007/s10753-019-01153-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-019-01153-4