
Abstract
Respiratory syncytial virus (RSV) is the leading cause of acute lower respiratory tract infection (LRTI) in children under 5 years of age, especially infants with severe bronchiolitis. Our preliminary clinical experiments showed that bacterial colonization was commonly observed in children with virus-induced wheezing, particularly in those with recurrent wheezing, suggesting that bacterial colonization with an accompanying viral infection may contribute to disease severity. In most cases, RSV-infected infants were colonized with pathogenic bacteria (mainly Gram-negative bacteria). LPS is the main component of Gram-negative bacteria and acts as a ligand for Toll-like receptor 4 (TLR4). Relevant studies have reported that the TLR family is crucial in mediating the link between viral components and immunologic responses to infection. Of note, TLR4 activation has been associated with disease severity during RSV infection. In the present study, we identified that LPS aggravated RSV-induced AHR and airway inflammation in BALB/c mice using an RSV coinfection model. We found that the airway inflammatory cells and cytokines present in BALF and TRIF in lung tissue play a role in inducing AHR and airway inflammation upon RSV and bacteria coinfection, which might occur through the TRIF-MMP-9-neutrophil-MMP-9 signalling pathway. These results may aid in the development of novel treatments and improve vaccine design.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Respiratory syncytial virus (RSV) is the leading cause of acute lower respiratory tract infection (LRTI) in children under 5 years of age [1, 2], especially in infants with severe bronchiolitis, who require hospitalization and are at increased risk of developing recurrent wheezing or childhood asthma [3,4,5]. Cooper BS et al. [6] reported that coinfection of bacteria and viruses had a synergistic effect, leading to more severe disease and hospitalization. It was also reported that up to 40% of children with severe RSV bronchiolitis requiring admission to the PICU were coinfected with bacteria in their lower airways and were at increased risk for bacterial pneumonia [7]. Our preliminary clinical experiments showed that bacterial colonization was common in children with virus-induced wheezing, particularly those with recurrent wheezing, suggesting that bacterial and viral coinfection may contribute to the severity of wheezing [8]. Bacterial infection is frequent in preschool children with persistent wheezing, and NTHi (Haemophilus influenzae), Streptococcus pneumonia, and Moraxella catarrhalis are the main pathogens involved in this process [9]. More frequently, RSV-infected infants are colonized with pathogenic bacteria and have a higher proportion of Gram-negative bacterial colonization compared to healthy age-matched controls [10].
Several studies have reported that RSV infection is often associated with secondary bacterial [11] or enhanced bacterial colonization of the respiratory epithelium, which permits microorganisms to overcome physical barriers to infection and evade innate immune responses [12]. An important family of immune-modulating receptor molecules that might be crucial to mediating the linkage of viral components and immunologic responses is the Toll-like receptor (TLR) family of immune-recognition receptors. Particularly, the role of TLR4 has been extensively studied in recent years. TLR-4 contributes to immune reorganization of RSV, and the ability to up-regulate TLR4 is closely linked to disease severity [13, 14]. Our previous results showed that TLR4 plays a critical role in LPS-mediated IL-6 responses in RSV-infected epithelial cells and might be an important factor that influences the cytokine-chemokine profile of epithelial cells by interacting with virus and endotoxin, which is correlated with the phenotypes of RSV disease [15]. TLR family members typically bind evolutionary conserved structures of a wide range of pathogens that are essential for the survival of these pathogens (pathogen-associated molecular patterns) and include bacterial carbohydrates, N-formylmethionine, lipoproteins, and fungal glucans. Mainly, there are two TLR4 signalling pathways that lead to production of either interleukins, such as IL-6 and IL-8, or type I IFNs. The first pathway acts via myeloid differentiation primary response gene 88 (MyD88). The second pathway acts via the adaptor protein TICAM-1 (TIR domain containing adaptor molecule-1)/TRIF (TIR domain containing adaptor protein-inducing interferon α).
Matrix metalloproteinase-9 (MMP-9) is a member of a subgroup of zinc endopeptidases that has been identified to degrade basement membrane components, including collagen type IV, fibronectin, and gelatine. MMP-9 is therefore considered to be essential for PMN migration and alveolar capillary leakage. Therefore, MMP-9 inhibition is promising for reduction or prevention of lung impairment. In an influenza infection mouse model, MMP-9 expression is required for neutrophil migration, which is correlated with disease severity [16, 17]. Previous studies have shown that MMP-9 production occurs via the MyD88 pathway following focal cerebral ischemia [18]. In addition, RSV-infected infants colonized with Gram-negative bacteria have an increased concentration of plasma IL-6 [10].
Despite previous reports stating that bacterial colonization (mainly Gram-negative bacteria) and coinfection with virus may contribute to the severity of LRTI, there are few comprehensive studies on this subject. Thus, there is poor knowledge of the pathophysiology and a lack of efficient data of therapeutical interventions. LPS, which is the main component of Gram-negative bacteria, is a TLR4 ligand [10, 19]. In our pre-clinical study, we utilized an LPS- and RSV-superinfected mouse model to explore underlying pathogenesis. According to the effects of airway inflammation-associated cytokines (such as MMP-9, IL-6) in BALF and MyD88/TRIF in lung tissue, we examined the pathogenesis of RSV coinfection with Gram-negative bacteria. The findings from this study may assist in the development of novel treatments and improve vaccine design.
METHODS AND MATERIALS
RSV and LPS Preparation
A stock of human A2 strain RSV was propagated and titered as previously described [20]. Virus pools were aliquoted, quick-frozen on dry ice/alcohol, and stored at −80 °C. The supernatants of uninfected Hep-2 cells were generated under the same condition. LPS (Escherichia coli 055:B5, Sigma), was assayed using the limulus hemocyanin agglutination assay.
Animal and Inoculation Procedure
Adult 6- to 8-week-old female BALB/c mice were purchased from Chongqing Medical University Animal Laboratory. All animals were housed in sterile microisolator cages with sterile food and water provided ad libitum and were maintained according to the guidelines of the Animal Welfare Act. The institutional animal care and use committee approved all experimental procedures.
Mice were divided into four groups:
-
The control group: Mice were sham-infected under anaesthesia by intranasal inoculation with DMEM medium containing 2% FBS on day 0 and were then inoculated with 50 μl of PBS over the following 3 days in the same way.
-
The LPS group: Mice were sham-infected under anaesthesia by intranasal inoculation with DMEM medium containing 2% FBS on day 0 and were then inoculated with LPS (10 μg/50 μl PBS) over the following 3 days in the same way.
-
The RSV group: Mice were infected under anaesthesia by intranasal inoculation with RSV (107 PFU in 100 μl of virus supernatant) on day 0 and were then inoculated with 50 μl of PBS over the following 3 days in the same way.
-
The RSV + LPS group: Mice were infected under anaesthesia by intranasal inoculation with RSV on day 0 and were then inoculated with LPS (10 μg/50 μl PBS) over the following 3 days in the same way.
Our previous work demonstrated that mice assessed during the first 7 days post-RSV infection showed significant differences (including the severity of the disease parameters, inflammatory cellular types, and proinflammatory cytokines) in contrast to mice assessed during the later stages of infection (from day 14 on). Based on these observations, the acute phase of RSV infection was indicated as the first 7 days and the chronic phase was indicated as the time period after day 14 [21, 22]. To examine the acute and chronic phases simultaneously, we chose 5, 7, and 14 days post-RSV treatment for our studies.
Determination of the Viral Loads
Viral loads were determined by Taq-probe after RSV infection. Briefly, total RNA was extracted from whole-lung tissue, and then, cDNA was synthesized. Primers specific for the nucleocapsid (N) gene of RSV were used [23]. A real-time PCR instrument (Applied Biosystems) was used with the following conditions: 1 cycle at 50 °C for 2 min, 1 cycle at 95 °C for 10 min, 40 cycles at 95 °C for 15 s, and 1 cycle at 60 °C for 1 min. The RSV load values were expressed as the log10 copy number of RSV-RNA/ml. The RSV subtype A plasmid was used as a positive control. Negative controls and serial dilutions of the positive controls were included in every PCR assay.
Bronchoalveolar Lavage Fluid (BALF) Analysis
After functional airway measurements, mice were deeply anaesthetised by an i.p. injection of pentobarbital (90-mg/kg body weight). Then, sections of the abdominal vessels were obtained from mice. The lungs were lavaged, in situ, six times with 0.5 ml of ice-cold PBS. The BAL fluid was centrifuged (2500 rpm, 5 min). The supernatants were stored at −80 °C prior to cytokine profile characterization. The cellular pellets were resuspended in 1 ml of PBS, and the total cell number was counted. Data are shown as cells/ml BALF. For differential cell counts, 250 μl of the resuspended cells was spun onto microscope slides, air-dried, and stained (Giemsa). For each sample, 200 cells were counted for the number of macrophages, eosinophils, neutrophils, and lymphocytes.
Lung Histology
For histological analysis, lung tissue was removed and fixed immediately in 10% (vol/vol) neutral buffered formalin for more than 24 h, embedded in paraffin, cut into 5-μm thick sections, and stained with haematoxylin and eosin (H&E; Sigma). Lung sections were microscopically reviewed and semi-quantitatively assessed for the degree and distribution of inflammation by a pathologist who was blinded to the treatment conditions using a previously published scoring system [24]. Each inflammatory parameter was independently assigned a value from 0 to 3, with higher scores reflecting greater inflammatory changes in the lung.
Measurement of AHR
AHR was measured in mice using a whole-body plethysmography (Emca instrument; Allmedicus, France) as previously described [20]. After a brief acclimatization to the chamber, the mice received an initial baseline challenge of PBS followed by increasing doses of a methacholine solution (3.125, 6.25, 12.5, 25, and 50 mg/ml; Sigma) in PBS for 3-min exposures. Afterwards, bronchoconstriction was recorded for an additional 5-min post-methacholine treatment. AHR was expressed as an enhanced pause (Penh). Penh, a dimensionless parameter, which is used to measure pulmonary resistance, was calculated by the changes in the chamber pressure induced by methacholine challenge during inspiration and expiration. The Penh values are reported as an average.
Measurement of Inflammatory Mediators in BALF
The cytokine levels in BALF were measured by ELISA. IFN-γ, IL-6 (Sizhengbai, Beijing, China) and MMP-9 (R&D) were performed according to the manufacturer’s directions.
Western Blot Analysis
Total protein extracts from lung tissues were obtained by using a total protein extraction kit (KeyGEN, Nanjing, China), and the protein concentration was determined using BCA assay reagents (Bioteke) according to the manufacturer’s protocol. Lung protein extracts containing equal quantities of protein were separated on a 10% SDS-PAGE (sodium dodecyl sulphate-polyacrylamide gel electrophoresis) gel and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA). After blotting, the membranes were blocked in blocking buffer containing 5% non-fat dried milk in 0.1% Tween 20 for 1 h at room temperature prior to incubation overnight at 4 °C with primary antibodies (TRIF, Enzo Life Sciences; MyD88, Santa Cruz Biotechnology; β-actin, Abio, Beijing, China). Next, membranes were washed and sequentially incubated with rabbit anti-goat Ig (Minneapolis). Finally, antibody-labelled proteins were visualized using ECL (Bio-Rad) and an imaging system (Bio-Rad). Signals were quantified by use of Quantity One software (Bio-Rad, Hercules, CA) and normalized relative to β-actin.
Statistics
GraphPad Prism 5.0 software (GraphPad Software Inc., La Jolla, CA, USA) was used to analyse the data. Data were expressed as the mean ± SEM. Analysis of variance (ANOVA) was used to determine the level of significant differences among all groups. Groups were compared by using the unpaired t test. The P value for significance was set to 0.05.
RESULTS
RSV Replication in a Murine Infection Model
To examine RSV replication in RSV-infected mice, lung tissue was collected on 5, 7, and 14 days post-RSV treatment. As shown in Fig. 1, on day 5, there was a significant decrease in the gene copy number for the RSV + LPS group compared with the RSV group (P < 0.001). There were no differences between the two groups on days 7 and 14. Over time, the RSV gene copy number decreased.

Viral copy numbers of RSV infection mice in lung tissues (log10 copy number of RSV-RNA/mL). The results are expressed as the mean ± S.E.M. the RSV + LPS group vs. the RSV group: @@@ P < 0.001. n = 4–5/group.
The Interaction of LPS and RSV Infection Increased the Inflammatory Reaction
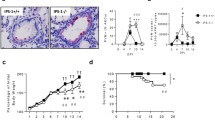
To evaluate the effect of coinfection of LPS and RSV on inflammatory reactions in the lung, total and differential cell counts in the BALF were measured. On days 5, 7, and 14, the total number of cells of the RSV + LPS group was higher than those of the RSV (P < 0.05) and LPS groups (P < 0.001) (Fig. 2A). On days 5, 7, and 14, the lymphocyte number of the RSV + LPS group was lower than that of the RSV group (P < 0.001, P < 0.001, P < 0.05) (Fig. 2C). On days 5, 7, and 14, the neutrophil number of the RSV + LPS group was higher than that of the LPS group (P < 0.05, P < 0.001, P < 0.05) (Fig. 2B). On days 5, 7, and 14, the macrophage number of the RSV + LPS group was higher than those of the RSV (P < 0.05, P < 0.05, P < 0.01) and LPS groups (P < 0.05, P < 0.05, P < 0.01) (Fig. 2D).

Total cell number and differential cell number in BALF. The results are expressed as the mean ± S.E.M. vs. control: *P < 0.05. **P < 0.01. ***P < 0.001. The RSV + LPS group vs. the RSV group: @ P < 0.05. @@@ P < 0.001. n = 7/group.
Lungs of infected mice were analysed using histopathology. Inflammation around the bronchioles and perivascular tissue as well as alveolitis in the lung were examined at days 5, 7, and 14 post-infection. Compared to the control group, there was an overall dramatic increase of lung inflammation in the LPS, RSV, and RSV + LPS groups on days 5 and 7. On day 14, mononuclear cell recruitment in the bronchioles and perivascular tissue as well as alveolitis of the RSV + LPS group were still increased, while they were decreased in the LPS and RSV groups (Fig. 3).

Histopathological changes of mouse lung (H&E staining, ×200). The results are expressed as the mean ± S.E.M. vs. control: ***P < 0.001. The RSV + LPS group vs. the RSV group: @ P < 0.05. @@@ P < 0.001. n = 3/group.
The Interaction of LPS and RSV Infection Increased the AHR
AHR was evaluated by a Penh value calculation (enhanced pauses) at 5, 7, and 14 days after RSV infection. On day 5, the Penh values for the LPS, RSV, and RSV + LPS groups were higher than those of the control at a methacholine concentration of 50 mg/m (P < 0.001), and no significant difference was identified among the three groups (Fig. 4A). On day 7, the Penh value of the LPS was comparable to that of the control group. The Penh value for the RSV group was higher than that of the control at a methacholine concentration of 50 mg/m (P < 0.001). Meanwhile, the Penh value for the RSV + LPS group was higher than that of the control at methacholine concentrations of 0, 3.125, 6.25, 12.5, 25, and 50 (P < 0.05, P < 0.01, P < 0.01, P < 0.01, P < 0.05, P < 0.001). There was no significant difference between the RSV and RSV + LPS groups (Fig. 4B). On day 14, the Penh value for the RSV group was higher than that of the control group at methacholine concentrations of 50 mg/m (P < 0.01). In addition, the Penh value of the RSV + LPS group was higher than that of the control at methacholine concentrations of 25 and 50 mg/m (P < 0.01, P < 0.001). The Penh value of the RSV + LPS group was higher than that of the RSV group at methacholine concentration of 25 and 50 mg/ml (P < 0.05, P < 0.001) (Fig. 4C).

Expiratory resistance methacholine challenge for all groups (Penh). The results are expressed as the mean ± S.E.M. vs. control: *P < 0.05. **P < 0.01. ***P < 0.001. The RSV + LPS group vs. the RSV group: @ P < 0.05. @@@ P < 0.001. n = 10/group.
The Interaction of LPS and RSV Infection Changed Cytokines
To evaluate cytokine profile changes, the presence of IFN-γ, MMP9, and IL-6 in the BALF was investigated. ELISA-based assays were used to determine the IFN-γ, MMP9, and IL-6 concentrations at 5, 7, and 14 days post-RSV infection. On day 5, production of IFN-γ in the RSV and RSV + LPS groups was significantly increased compared to that of the control group (P < 0.001, P < 0.001). The concentrations of IFN-γ between the RSV and RSV + LPS groups were almost equivalent on day 5 (Fig. 5A). On day 7, production of IFN-γ in the RSV group was significantly higher than that of the control group, but was decreased in the RSV + LPS group (P < 0.05, P ≥ 0.05) (Fig. 5B). On day 5, the production of MMP-9 in the LPS and RSV + LPS group was significantly increased compared to the control group (P < 0.001, P < 0.001). The concentration of MMP-9 in RSV + LPS group was higher than that of the RSV group on day 5 (P < 0.001) (Fig. 5D). On day 7, production of MMP-9 in the RSV + LPS group was still significantly higher than that of the control group, yet it was decreased in the LPS group (P < 0.05, P ≥ 0.05) (Fig. 5E). On days 5, 7, and 14, production of IL-6 in all groups was similar (Fig. 5G, H, and I). On day 14, production of IFN-γ and MMP-9 in both groups was reduced (Fig. 5C and F).

Cytokine concentration in BALF. The results are expressed as the mean ± S.E.M. vs. control: *P < 0.05. **P < 0.01. ***P < 0.001. The RSV + LPS group vs. the RSV group: @ P < 0.05. @@@ P < 0.001. n = 4/group.
The Relative Expression of TRIF/MyD88 in Different Groups of Mice
We next detected the relative expression of TRIF and MyD88 in lung tissue. We found that the relative expression of TRIF in the RSV + LPS group was significantly higher than that of the control group (P < 0.01). Moreover, increased expression of TRIF in the LPS and RSV groups was not evident compared to that of the control group. There was no change in My88 expression in each group studied (Fig. 6).

Expression of TRIF and MyD88 in lung tissues. The results are expressed as the mean ± S.E.M. vs. control: *P < 0.01. n = 3.
DISCUSSION
In this study, we utilized the LPS and RSV coinfection model to investigate the role of LPS in aggravating AHR and airway inflammation in BALB/c mice. In addition, we found that airway inflammatory cells and cytokines present in BALF and TRIF in lung tissue play a role in this reaction. We conclude that TRIF-MMP-9-neutrophil-MMP-9 might be the main signalling pathway by which the interaction of LPS and RSV occurs.
We analysed the types of inflammatory cells in BALF. The results indicated that neutrophils significantly contribute to airway inflammation in the RSV + LPS group. Infection of the airway mucosa with inflammatory cells was thought to be an important factor in the pathogenesis of a broad spectrum of airway disorders. As first line defence cells, rapid recruitment and efficient activation of PMNs are of critical importance. Lymphocytes were shown to be the primary inflammatory cells in the RSV group and neutrophils in the LPS group. Furthermore, we found that neutrophils were the main inflammatory cell type detected in the RSV + LPS group, which is consistent with previous studies [25, 26] performed in severe RSV-infected children and mice. Colonization by pathogenic bacteria was associated with higher blood neutrophil percentages and a greater number of nasal wash WBC, especially in those colonized with Gram-negative bacteria [10]. Neutrophils may play a crucial role in this process in the following ways: (1) Neutrophils play a vital role in innate immune reactions and contribute to the immune response of the airway; (2) neutrophils can cause tissue damage via production and release of oxygen radicals, proteases, and soluble mediators of inflammation (e.g., cytokines and chemokines); and (3) neutrophils are involved in airway remodelling, which eventually leads to an enhanced response to MCh [27]. It had been demonstrated that neutrophil-mediated inflammation is involved in the pathogenesis of tissue destruction and augmentation of bronchial reactivity in RSV [19]. All of these data indicate that neutrophils play an important role in the copathogenesis of RSV and bacteria-infected mice.
We examined the inflammatory cytokines in BALF and showed that MMP-9 was expressed at higher levels in the RSV + LPS group compared to the LPS and RSV groups. MMP-9 is essential for PMN migration and pivotal to the initiation of inflammation [26, 28]. Similarly, our results revealed a relationship between MMP-9 and neutrophils. Hence, MMP-9 inhibition is a promising target for reduction or prevention of lung impairment. Increased MMP-9 activity had been demonstrated in BALF following CPB in a swine model [29]. These findings suggest that MMP-9 plays an important role in RSV-infected mice colonized with Gram-negative bacteria. Moreover, our results show that LPS shifts the expression of the inflammatory cytokine IFN-γ to MMP-9 in RSV infected mice, which may explain why RSV-infected mice colonized with Gram-negative bacteria display high AHR and airway inflammation.
Our preliminary work demonstrated high levels of TRIF expression in RSV-infected mice [20]. In this study, expression of TRIF in the RSV group was also higher than that in the control group. In addition, TRIF expression was higher in the RSV + LPS group compared to the RSV group. At the same time, there was no change in MyD88 expression in all groups tested, which is inconsistent with our new study, which showed that the SARM-TRIF-signalling pathway is involved in regulating MMP-12 overproduction [21].
Neutrophils can cause tissue damage via production and release of oxygen radicals, proteases, and soluble mediators of inflammation (e.g., cytokines and chemokines) [27]. The rapid recruitment and efficient activation of neutrophils as the first line of defence are of critical importance. MMP-9 is considered to be essential for PMN migration and plays a pivotal role in the initiation of inflammation [26, 28]. Neutrophil-mediated migration to the respiratory tract in response to influenza infection requires MMP-9 and is dependent on extrinsic TLR-signalling [30]. Therefore, MMP-9 is considered to be essential for PMN migration and alveolar capillary leakage. Moreover, MMP-9 inhibition is a promising target for the reduction or prevention of lung impairment. We speculated that TRIF-MMP-9-neutrophils might be the main pathway utilized by bacteria upon RSV coinfection in mice. McNamara PS et al. [31] reported that MMP-9 expression was present at high concentrations in neutrophils, which are the major immune cells present in human BALF from RSV patients. In combination with our results that showed that neutrophils, TRIF, and MMP-9 were higher in the RSV + LPS groups, we conclude that TRIF-MMP-9-neutrophil-MMP-9 might be the main pathway utilized by bacteria upon RSV coinfection in mice.
In our experiment, the viral load of RSV in the RSV group was not lower than that of the RSV + LPS group. In other words, LPS did not increase AHR and airway inflammation by up-regulating the viral load, which does not conform with existing results. Previous work has shown a direct correlation between a high viral load and increased disease severity in patients coinfected with HBoV1, which suggests that HBoV1 could cause LRTIs, but a symptomatic HBoV infection was only observed in the context of a high viral load [32]. A high load of HRV-A in the lower respiratory tract may be connected with disease severity in children younger than 2 years [33]. Disease severity correlated positively with viral load during primary RSV infection [34]. However, other studies have reported conflicting results, which may be confounded by the presence of risk factors, such as cardiopulmonary disease, by RSV immunization or by the occurrence of coinfections since none of the mentioned studies considered copathogens during RSV infection. Hence, we speculate that LPS changes the viral load of RSV. Previous reports have shown that TLR4-deficient mice challenged with RSV exhibit impaired viral clearance compared to mice expressing TLR4. These findings suggest that Toll-like signalling pathways play an important role in innate immunity to RSV [35]. LPS, a TLR4 specific agonist, could stimulate TLR4 to promote viral clearance and may be the reason that the viral load of RSV in the RSV + LPS group was lower than that of the RSV group. The RSV gene copy number decreased over time, so the difference in viral load between the two groups disappeared. Our results were completely in agreement with Caballero’s report, which showed that there was no association between the severity of RSV disease and viral titers detected in respiratory secretions [36].
In conclusion, the interaction of RSV and bacteria functions to induce AHR and airway inflammation, possibly through the TRIF-MMP-9-neutrophil-MMP-9 signalling pathway. In future studies, we plan to use specific blockers of this interaction to explore the direct relationship between cytokine expression and airway inflammation.
References
Collins, P.L., and J.A. Melero. 2011. Progress in understanding and controlling respiratory syncytial virus: still crazy after all these years. Virus Research 162 (1–2): 80–99. doi:10.1016/j.virusres.2011.09.020.
Escobar, Gabriel J., Anthony S. Masaquel, Sherian X. Li, Eileen M. Walsh, and Patricia Kipnis. 2013. Persistent recurring wheezing in the fifth year of life after laboratory-confirmed, medically attended respiratory syncytial virus infection in infancy. BMC Pediatrics 13 (1): 97.
Wu, P., and T.V. Hartert. 2011. Evidence for a causal relationship between respiratory syncytial virus infection and asthma. Expert Review of Anti-Infective Therapy 9 (9): 731–745. doi:10.1586/eri.11.92.
Bacharier, Leonard B., Rebecca Cohen, Toni Schweiger, Huiquing Yin-DeClue, Chandrika Christie, Jie Zheng, Kenneth B. Schechtman, Robert C. Strunk, and Mario Castro. 2012. Determinants of asthma after severe respiratory syncytial virus bronchiolitis. Journal of Allergy and Clinical Immunology 130 (1): 91–100. e103.
Sigurs, Nele, Fatma Aljassim, Bengt Kjellman, Paul D. Robinson, Fridrik Sigurbergsson, Ragnar Bjarnason, and Per M. Gustafsson. 2010. Asthma and allergy patterns over 18 years after severe RSV bronchiolitis in the first year of life. Thorax 65 (12): 1045–1052.
Cooper, Ben S., Daniel M. Weinberger, Keith P. Klugman, Claudia A. Steiner, Lone Simonsen, and Cécile Viboud. 2015. Association between respiratory syncytial virus activity and pneumococcal disease in infants: a time series analysis of US hospitalization data. PLoS Medicine 12 (1): e1001776. doi:10.1371/journal.pmed.1001776.
Thorburn, K., S. Harigopal, V. Reddy, N. Taylor, and H.K. van Saene. 2006. High incidence of pulmonary bacterial co-infection in children with severe respiratory syncytial virus (RSV) bronchiolitis. Thorax 61 (7): 611–615. doi:10.1136/thx.2005.048397.
Yu, Deng, Liu Wei, Zhengxiu Luo, Jian Luo, Lijia Wang, Xiqiang Yang, Zhao Xiaodong, Fu Zhou, and Liu Enmei. 2010. Impact of bacterial colonization on the severity, and accompanying airway inflammation, of virus-induced wheezing in children. Clinical Microbiology and Infection 16 (9): 1399–1404.
De Schutter, I., A. Dreesman, O. Soetens, M. De Waele, F. Crokaert, J. Verhaegen, D. Pierard, and A. Malfroot. 2012. In young children, persistent wheezing is associated with bronchial bacterial infection: a retrospective analysis. BMC Pediatrics 12: 83. doi:10.1186/1471-2431-12-83.
Suarez-Arrabal, M.C., C. Mella, S.M. Lopez, N.V. Brown, M.W. Hall, S. Hammond, W. Shiels, et al. 2015. Nasopharyngeal bacterial burden and antibiotics: Influence on inflammatory markers and disease severity in infants with respiratory syncytial virus bronchiolitis. The Journal of Infection 71 (4): 458–469. doi:10.1016/j.jinf.2015.06.010.
Avadhanula, Vasanthi, Yan Wang, Allen Portner, and Elisabeth Adderson. 2007. Nontypeable Haemophilus influenzae and Streptococcus pneumoniae bind respiratory syncytial virus glycoprotein. Journal of Medical Microbiology 56 (9): 1133–1137.
Avadhanula, V., C.A. Rodriguez, J.P. Devincenzo, Y. Wang, R.J. Webby, G.C. Ulett, and E.E. Adderson. 2006. Respiratory viruses augment the adhesion of bacterial pathogens to respiratory epithelium in a viral species- and cell type-dependent manner. Journal of Virology 80 (4): 1629–1636. doi:10.1128/JVI.80.4.1629-1636.2006.
Gagro, Alenka, Mirna Tominac, Vilka Kršulović-Hrešić, Ana Baće, Mladen Matić, Vladimir Draženović, Gordana Mlinarić-Galinović, Ela Kosor, Katja Gotovac, and Ivan Bolanča. 2004. Increased Toll-like receptor 4 expression in infants with respiratory syncytial virus bronchiolitis. Clinical & Experimental Immunology 135 (2): 267–272.
Jorquera, Patricia A., Katie E. Oakley, and Ralph A. Tripp. 2013. Advances in and the potential of vaccines for respiratory syncytial virus. Expert Review of Respiratory Medicine 7 (4): 411–427.
Xie, Xiao-Hong, Helen K.W. Law, Li-Jia Wang, Xin Li, Xi-Qiang Yang, and En-Mei Liu. 2009. Lipopolysaccharide induces IL-6 production in respiratory syncytial virus-infected airway epithelial cells through the toll-like receptor 4 signaling pathway. Pediatric Research 65 (2): 156–162.
Kong, M.Y., J.P. Clancy, N. Peng, Y. Li, T.J. Szul, X. Xu, R. Oster, et al. 2014. Pulmonary matrix metalloproteinase-9 activity in mechanically ventilated children with respiratory syncytial virus. The European Respiratory Journal 43 (4): 1086–1096. doi:10.1183/09031936.00105613.
Bradley, L.M., Douglass, M.F., Chatterjee, D., Akira, S.,and Baaten, B.J.. 2012. Matrix metalloprotease 9 mediates neutrophil migration into the airways in response to influenza virus-induced toll-like receptor signaling. PLoS pathogens 8 (4).
Famakin, B.M., Y. Mou, K. Johnson, M. Spatz, and J. Hallenbeck. 2014. A new role for downstream Toll-like receptor signaling in mediating immediate early gene expression during focal cerebral ischemia. Journal of Cerebral Blood Flow and Metabolism 34 (2): 258–267. doi:10.1038/jcbfm.2013.182.
Sethi, S., J. Maloney, L. Grove, C. Wrona, and C.S. Berenson. 2006. Airway inflammation and bronchial bacterial colonization in chronic obstructive pulmonary disease. American Journal of Respiratory and Critical Care Medicine 173 (9): 991–998. doi:10.1164/rccm.200509-1525OC.
Zang, N., X. Xie, Y. Deng, S. Wu, L. Wang, C. Peng, S. Li, K. Ni, Y. Luo, and E. Liu. 2011. Resveratrol-mediated gamma interferon reduction prevents airway inflammation and airway hyperresponsiveness in respiratory syncytial virus-infected immunocompromised mice. Journal of Virology 85 (24): 13061–13068. doi:10.1128/JVI.05869-11.
Long, X., S. Li, J. Xie, W. Li, N. Zang, L. Ren, Y. Deng, et al. 2015. MMP-12-mediated by SARM-TRIF signaling pathway contributes to IFN-gamma-independent airway inflammation and AHR post RSV infection in nude mice. Respiratory Research 16: 11. doi:10.1186/s12931-015-0176-8.
Zang, Na, Simin Li, Wei Lia, Xiaohong Xie, Luo Ren, Xiaoru Long, Jun Xie, Yu Deng, Zhou Fu, Fadi Xu, and Liu Enmei. 2015. Resveratrol suppresses persistent airway inflammation and hyperresponsivess might partially via nerve growth factor in respiratory syncytial virus-infected mice. International Immunopharmacology 28(1): 8
Hu, A., M. Colella, J.S. Tam, R. Rappaport, and S.M. Cheng. 2003. Simultaneous detection, subgrouping, and quantitation of respiratory syncytial virus a and B by real-time PCR. Journal of Clinical Microbiology 41 (1): 149–154. doi:10.1128/jcm.41.1.149-154.2003.
Peebles, R. Stokes, James R. Sheller, Robert D. Collins, A. Kasia Jarzecka, Daphne B. Mitchell, Robert A. Parker, and Barney S. Graham. 2001. Respiratory syncytial virus infection does not increase allergen-induced type 2 cytokine production, yet increases airway hyperresponsiveness in mice. Journal of Medical Virology 63 (2): 178–188.
de Steenhuijsen Piters, W.A., Heinonen, S., Hasrat, R., Bunsow, E., Smith, B., Suarez-Arrabal, M.C., Chaussabel, D., Cohen, D.M., Sanders, E.A., Ramilo, O., Bogaert, D.,and Mejias, A.. 2016. Nasopharyngeal microbiota, host transcriptome and disease severity in children with respiratory syncytial virus infection. Am J Respir Crit Care Med.
Stoppelenburg, A.J., V. Salimi, M. Hennus, M. Plantinga, Veld R. in’t Huis, J. Walk, J. Meerding, F. Coenjaerts, L. Bont, and M. Boes. 2013. Local IL-17A potentiates early neutrophil recruitment to the respiratory tract during severe RSV infection. PloS One 8 (10): e78461. doi:10.1371/journal.pone.0078461.
Savov, J.D., S.H. Gavett, D.M. Brass, D.L. Costa, and D.A. Schwartz. 2002. Neutrophils play a critical role in development of LPS-induced airway disease. American Journal of Physiology. Lung Cellular and Molecular Physiology 283 (5): L952–L962. doi:10.1152/ajplung.00420.2001.
Puthothu, B., M. Krueger, J. Heinze, J. Forster, and A. Heinzmann. 2006. Impact of IL8 and IL8-receptor alpha polymorphisms on the genetics of bronchial asthma and severe RSV infections. Clin Mol Allergy 4: 2. doi:10.1186/1476-7961-4-2.
Wang, Changtian, Demin Li, Yajun Qian, Jun Wang, and Hua Jing. 2012. Increased matrix metalloproteinase-9 activity and mRNA expression in lung injury following cardiopulmonary bypass. Laboratory Investigation 92 (6): 910–916. doi:10.1038/labinvest.2012.50.
Bradley, L.M., M.F. Douglass, D. Chatterjee, S. Akira, and B.J. Baaten. 2012. Matrix metalloprotease 9 mediates neutrophil migration into the airways in response to influenza virus-induced toll-like receptor signaling. PLoS Pathogens 8 (4): e1002641. doi:10.1371/journal.ppat.1002641.
McNamara, P.S., P. Ritson, A. Selby, C.A. Hart, and R.L. Smyth. 2003. Bronchoalveolar lavage cellularity in infants with severe respiratory syncytial virus bronchiolitis. Archives of disease in childhood. 88 (10): 6.
Sadik, C.D., N.D. Kim, and A.D. Luster. 2011. Neutrophils cascading their way to inflammation. Trends in Immunology 32 (10): 452–460. doi:10.1016/j.it.2011.06.008.
Xiao, Q., S. Zheng, L. Zhou, L. Ren, X. Xie, Y. Deng, D. Tian, Y. Zhao, Z. Fu, T. Li, A. Huang, and E. Liu. 2015. Impact of human rhinovirus types and viral load on the severity of illness in hospitalized children with lower respiratory tract infections. The Pediatric Infectious Disease Journal 34 (11): 6.
Houben, M.L., F.E. Coenjaerts, J.W. Rossen, M.E. Belderbos, R.W. Hofland, J.L. Kimpen, and L. Bont. 2010. Disease severity and viral load are correlated in infants with primary respiratory syncytial virus infection in the community. Journal of Medical Virology 82 (7): 1266–1271.
Haynes, L.M., D.D. Moore, E.A. Kurt-Jones, R.W. Finberg, L.J. Anderson, and R.A. Tripp. 2001. Involvement of toll-like receptor 4 in innate immunity to respiratory syncytial virus. Journal of Virology 75 (22): 10730–10737. doi:10.1128/JVI.75.22.10730-10737.2001.
Caballero, M.T., M.E. Serra, and P.L. Acosta. 2015. TLR4 genotype and environmental LPS mediate RSV bronchiolitis through Th2 polarization. The Journal of Clinical Investigation 125 (2): 571–582. doi:10.1172/jci75183ds1.
Acknowledgments
This work is supported by the fund of the National Natural Science Foundation of China (Nos. 81501742 and 81470208).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zhou, N., Li, W., Ren, L. et al. An Interaction of LPS and RSV Infection in Augmenting the AHR and Airway Inflammation in Mice. Inflammation 40, 1643–1653 (2017). https://doi.org/10.1007/s10753-017-0604-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-017-0604-7