
Abstract
This study was undertaken to clarify the effects of urinary trypsin inhibitor (UTI) on lipopolysaccharide (LPS)-induced acute lung injury. Rabbits were randomly assigned to one of seven groups: saline only, UTI, LPS, pre- or post-UTI-high (infusion of UTI of 25,000 U/kg followed by 25,000 U/kg over 2 h), pre- or post-UTI-low (infusion of UTI of 2,500 U/kg followed by 2,500 U/kg over 2 h). UTI was administered 30 min before (pre-groups) or 15 min after (post-groups) LPS administration. Rabbits were mechanically ventilated with 40% oxygen for 6 h. LPS decreased peripheral blood leukocyte counts and increased wet/dry weight ratio of lung, lung injury score, neutrophil infiltration in lung, and IL-8 production in systemic blood and bronchoalveolar lavage fluid (BALF). Rabbits treated by UTI were protected from LPS-induced lung injury, as determined by wet/dry weight ratio, neutrophil infiltration in lung, lung injury score, and IL-8 in BALF levels. UTI attenuated LPS-induced acute lung injury in rabbits mainly by inhibiting neutrophil and IL-8 responses, which may play a central role in sepsis-related lung injury.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
Acute lung injury (ALI) is a complex syndrome of intense pulmonary inflammatory response with high morbidity and mortality and is characterized by increased production of proinflammatory cytokines, neutrophil accumulation, interstitial edema, disruption of epithelial integrity, and leakage of protein into the alveolar space [1]. The most common cause of ALI is well known to be sepsis [2, 3], but the precise pathogenesis for sepsis-induced ALI is not yet fully defined. However, massive accumulation of neutrophils in the lung and increased pulmonary proinflammatory cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin (IL)-1, IL-6, and IL-8 are major features of ALI.
Lipopolysaccharide (LPS), a major component of the outermost membrane of gram-negative (G(−)) bacteria [4], is the key mediator in the development of sepsis caused by G(−) bacteria [5] and induces the production of numerous proinflammatory mediators such as IL-8 [6]. IL-8 has been well known to the main chemotactic factor for neutrophils in the lung of patients with ALI [7]. It also has been known that the early appearance of IL-8 in BALF is an important prognostic indicator for the development of ALI [8]. Increased levels of IL-8 in patients with sepsis and ALI correlate to poor outcomes [9].
Urinary trypsin inhibitor (UTI) is a heat- and acid-stable glycoprotein and one of the kunitz-type serine protease inhibitors found in human urine and blood [10]. UTI inhibits inflammatory proteases, including trypsin, chymotrypsin, plasmin, cathepsin G, and leukocyte elastase [11]. Because UTI has a nature of multiple protease inhibition, UTI appears to prevent organ injury by inhibiting the activity of these serine proteases [12]. In addition, as with other serine type protease inhibitors, UTI has been reported to have anti-inflammatory properties besides being a protease inhibitor. UTI inhibits the enhanced production of proinflammatory mediators such as IL-8, TNF-α, and thromboxane B2 induced by LPS [13–15]. Inoue et al. reported that LPS-treated UTI knockout mice showed more severe neutrophilic inflammation in the lung and kidney, and more prominent expression of proinflammatory mediator in multiple organs compared with LPS-treated wild-type mice [16, 17]. These studies have indicated that proteases are critical mediators in LPS-induced organ injury.
The aim of the present study was to evaluate whether these beneficial effects of UTI improve LPS-induced acute lung injury in rabbits.
MATERIALS AND METHODS
Materials and Animals
Urinary trypsin inhibitor was a generous gift from Hanlim Pharmaceutical (Seoul, Korea). Immunoreactive IL-8 was quantified for human blood using commercially available ELISA kits (R&D System, Minneapolis, MN, USA). Escherichia coli 055:B5 endotoxin was purchased from Sigma-Aldrich (St. Louis, MO, USA). Male albino rabbits, weighing 2.2–2.8 kg, were purchased from Damul Science (Daejeon, Korea). The rabbits were kept on a 12-h light/dark cycle with free access to food and water. All experiments were conducted in accordance with the institutional review board-approved protocols.
Animal Preparation
Rabbits were initially anesthetized with ketamine hydrochloride (30 mg/kg, IM) and xylazin hydrochloride (0.3 mg/kg, IM). Intravenous angiocatheters were inserted into both ear veins for the route of the administration of fluids and drugs. Lactated Ringer’s solution was infused at a rate of 4 ml/kg/h until the end of the study. Tracheostomy was performed aseptically and a 3.5-mm uncuffed endotracheal tube was inserted into the trachea under spontaneous ventilation. After the start of continuous infusion of ketamine (3 mg/kg/h) and vecuronium bromide (0.05 mg/kg/h) for maintenance of anesthesia and paralysis of muscle, the lung of the rabbit was mechanically ventilated with 40% oxygen using a pressure controlled ventilator (Servo 900B, Siemen-Elema, Solna, Sweden). Inspiratory pressure and positive end expiratory pressure were set to 16 and 3 cmH2O, respectively. Respiratory rate was controlled to produce an initial arterial carbon dioxide tension of 35–45 mmHg. The rabbits were placed on a heating pad under a radiant heating lamp to keep body temperature between 36.5°C and 37.5°C at the esophagus. The arterial catheter was placed in the aorta via carotid artery cut-down to harvest blood samples.
Experimental Protocols
After the baseline measurements, animals were randomly assigned to one of seven groups: control group receiving intravenous infusion of saline only or UTI (Saline-Only group, n = 7 and UTI-Only group, n = 7, respectively); LPS control group (LPS-Only group, n = 7) receiving intravenous infusion of saline and 5 mg/kg E. coli endotoxin O55:B5; low-dose UTI treated group receiving intravenous infusion of UTI of 2,500 U/kg followed by 2,500 U/kg over 2 h (pre-UTI-Low group, n = 7 and post-UTI-Low group, n = 7, respectively); high-dose UTI treated group receiving intravenous infusion of UTI of 25,000 U/kg followed by 25,000 U/kg over 2 h (pre-UTI-High group, n = 7 and post-UTI-High group, n = 7, respectively). UTI was administered 30 min before (pre-groups) or 15 min after (post-groups) LPS administration. The Saline-, UTI-, and LPS-Only groups were infused with an equivalent volume of saline instead of LPS or UTI. The basal infusion of Lactated Ringer’s solution (4 ml/kg/h) was started 0.5 h before saline or LPS administration (Fig. 1). Systolic arterial pressure, arterial blood sample for blood cell counts, and blood gas analysis were obtained at 0, 1, 2, 3, 4, 5, and 6 h after saline or LPS administration. All rabbits were killed by injection of an overdose of thiopental sodium. The wet weight/dry weight (W/D) ratio of right lung and lung injury score of left lung, and number of leukocytes, percent of polymorphonuclear neutrophil (PMN) cells, and IL-8 in bronchoalveolar lavage fluid (BALF) were measured.

Experimental design. Animals were randomly assigned to one of seven groups. Saline-Only group received intravenous infusion of saline only. UTI- and LPS-Only groups received intravenous infusion of UTI (IV infusion of UTI of 25,000 U/kg followed by 25,000 U/kg over 2 h) and 5 mg/kg E. coli endotoxin O55:B5 respectively. Low-dose UTI treated group received intravenous infusion of UTI of 2,500 U/kg followed by 2,500 U/kg over 2 h (pre-UTI-Low group and post-UTI-Low group, respectively). High-dose UTI treated group received intravenous infusion of UTI of 25,000 U/kg followed by 25,000 U/kg over 2 h (pre-UTI-High group and post-UTI-High group, respectively). UTI was administered 30 min before (pre-groups) or 15 min after (post-groups) LPS administration. The Saline-, UTI-, and LPS-Only groups were infused with an equivalent volume of saline instead of LPS or UTI, respectively. S saline, E E. coli endotoxin O55:B5, UTI urinary trypsin inhibitor.
Peripheral Cell Counts
The numbers of peripheral leukocytes were measured with a cell counter (XE2100, Sysmex, Kobe, Japan).
Lung Wet Weight to Dry Weight (W/D) Ratio
The left upper lobe of the lung was weighed and then dried to constant weight at 60°C over 72 h in an oven. To assess tissue edema, the W/D ratio was calculated.
Histopathological Examination
The left lower bronchus was cannulated and pressure-inflated (20 cmH2O) with 10% formaldehyde solution. The bronchus then ligated and suspended for at least 24 h in formaldehyde solution prior to embedding in paraffin. Lung sections were stained with hematoxylin and eosin. Two observers, unaware of the nature of the experiment, scored the lung injury under light microscopy from 0 (no damage) to 4+ (severe damage), according to the combined assessment of infiltration/aggregation of neutrophils, alveolar congestion, hemorrhage, edema, thickness of the alveolar wall, and hyaline membrane formation in the airspace or vessel wall [18]. To count the number of neutrophils in the airspace, five randomly selected fields per slide were read at ×400 magnification by the pathologist. Fields containing large vessels or bronchi were excluded. The number of neutrophils per field was normalized to alveoli per field to control for inflation of the lung.
Preparation of Bronchoalveolar Lavage Fluid
The BALF was harvested from the right lung. Through the right mainstem bronchus, 35 ml of phosphate buffered saline (PBS) was infused slowly and withdrawn three times. The PBS contained ethylendiamine-tetraacetic acid-2Na and was cooled to 4°C to prevent metabolism of leukocytes. The BALF was analyzed for cell count and cell differentiation. A cytocentrifuged spin preparation (CF-RD, Sakura, Tokyo, Japan) of the BALF was stained with Wright-Giemsa for cell differentiation. The numbers of leukocytes in the BALF were counted with a cell counter (XE2100, Sysmex, Kobe, Japan). The fluid was then centrifuged at 3,500×g at 4°C for 20 min to remove the cells. The cell-free supernatant was stored at −80°C for measurements of IL-8.
Measurements of Mediators in Bronchoalveolar Lavage Fluid
Immunoreactive IL-8 was quantified using commercially available ELISA kits (R&D Systems, Minneapolis, MN, USA), according to the manufacturer’s instructions and as described previously. The assay kit cross-reacts with IL-8 and human recombinant IL-8 were used as the standard [19].
Statistical Analysis
Data from the experiments are expressed as a mean ± SD except the lung injury score which is given as a median. Data from three or more groups were compared using one-way analysis of variance followed by the Scheffé multiple comparison test. Pairwise comparisons were made with the Student’s t test. A value of p < 0.05 was considered significant.
RESULTS
Changes in Hemodynamics, Peripheral Blood Leukocyte Counts
The systolic arterial pressure and arterial oxygen partial pressure (data not shown) did not change significantly during the study period in all groups, but peripheral blood leukocyte counts decreased gradually in all groups treated with LPS compared with the Saline-Only group. However, the decrease of peripheral blood leukocyte counts was attenuated in pre-UTI-High group compared with LPS-Only group (p < 0.05; Table 1).
Analysis of Bronchoalveolar Lavage Fluid
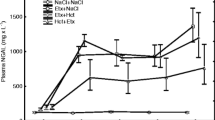
The average recovered BALF was about 75% in the seven groups. This range of percentages indicates a similar dilution among the animals. Total cell counts and neutrophil to total cell ratio were not different among seven groups (leukocyte counts, 370 ∼ 460/ul; PMNL to total cell count ratio, 2.5 ∼ 3.2%). In contrast, the concentration of IL-8 in the BALF significantly increased in LPS-Only group compared with the Saline-Only group. UTI attenuated the LPS-induced IL-8 production in BALF of groups treated with LPS except post-UTI-Low group (p < 0.05; Table 2, Fig. 2).

Effects of UTI on LPS-induced changes in the concentration of interleukin-8 in bronchoalveolar lavage fluid (BALF) and wet/dry weight (W/D) ratio. BALF and lung lobe were collected 6 h after the start of the saline or LPS infusion. Each value represents mean ± SD from seven rabbits. *P < 0.05 versus group Saline-Only, † P < 0.05 vs. group LPS-Only, ‡ P < 0.01 vs. group LPS-Only.
Lung Wet/Dry Weight Ratio
The W/D weight ratio increased in the LPS-Only group compared with the Saline-Only group. The increase of this ratio was attenuated in the pre-UTI-High group (p < 0.05; Table 2, Fig. 2).
Histopathologic Grading
Light microscopic findings in the LPS-Only group include edema, hemorrhage, thickening of the alveolar wall, and infiltration of inflammatory cells in lung parenchyma. Pre- and post-treatment with high-dose UTI attenuated this histopathologic severity of the lung injury induced by LPS (p < 0.05; Table 2, Fig. 3). LPS-induced neutrophils infiltration in lung was attenuated in the pre-UTI-High group (Table 3).

Effects of UTI on lung tissue damage in rabbits at 6 h after the start of saline or LPS infusion. Representative photomicrographies showing hematoxylin and eosin staining samples with median values in a group Saline-Only, b group LPS-Only, and c group pre-UTI-High. Original print magnifications ×200. Lung injury score d: lung injury was scored from 0 (no damage) to 4+ (severe damage) according to the criteria described in MATERIALS AND METHODS. Bars represent median values from seven rabbits. *P < 0.05 vs. group Saline-Only. † P < 0.05 vs. group LPS-Only.
DISCUSSION
Various inflammatory mediators, including neutrophils, cytokines/chemokines, lipid mediators (phospholipase A2, thromboxane A2, prostacyclin), platelet activating factor, and proteases contribute to the pathogenesis of LPS-induced acute lung injury [20]. Among them, the products from neutrophils are recognized to play important roles. Neutrophils stimulated by LPS generate neutrophil elastase, oxygen radicals, nitric oxide, and other mediators of inflammatory processes.
Pre-treated UTI attenuated LPS-induced acute lung injury, as determined by development of lung edema, IL-8 in BALF levels, neutrophils infiltration, and histopathologic findings in the lung, as shown in the present study. These findings are consistent with previous observation in which UTI inhibited both expression of neutrophil chemoattractant and infiltration of neutrophils in the lung [16, 17]. UTI attenuated the increases of IL-8 concentration of BALF in the rabbits treated with LPS, which is an important finding because IL-8 is associated with the prognosis of ALI. In trauma patients, the early appearance of interleukin-8 in BALF is an important prognostic indicator for the development of ARDS [8]. Increased levels of proinflammatory cytokines in patients with sepsis and ALI, including IL-8 and IL-6, correlate to a poor outcome [9].
In the present study, interestingly, UTI reduced the production of potent chemoattractant IL-8 in BALF, but not in systemic circulation. However, LPS-induced neutrophils infiltration of lung was attenuated in the Pre-UTI high group. Previous studies reported that UTI inhibits LPS-induced production of IL-8 in vascular endothelial cells [21], neutrophil elastase-induced IL-8 gene expression [22, 23], expression of intercellular adhesion molecule-1, endothelial cell adhesion molecule-1 on endothelial cells, and transendothelial migration of neutrophils stimulated by IL-8 [24]. Also, UTI not only inactivated elastase secreted by neutrophils, but also suppressed the production and secretion of elastase from neutrophils which play an important role of diapedesis and extravasation of granulocytes [21, 25, 26]. Even though we did not prove directly that UTI decreases the production of IL-8 in specific cells such as macrophage or epithelial cell and decreases migration of neutrophils (chemotatic effects) into lung, it is likely that UTI attenuated the histopathologic severity of lung parenchyma through the inhibition of neutrophil migration into the lung parenchyma as shown in our study.
In direct insult models, the injured structures are primarily focused on the alveolar epithelium, which leads to activation of local macrophages, neutrophils, and proinflammatory cytokines. But, in indirect pulmonary injury, the first site of damage is the pulmonary vascular endothelium by inflammatory mediator released from extrapulmonary foci into the systemic circulation. Although various causes of ALI result in similar pathologies in the late stage [1] and the distinction between pulmonary and extrapulmonary ALI is also not always clear and simple, indirect insult appear to spare relatively the intra-alveolar spaces and alveolar epithelium at the early phase [27]. In this study, UTI attenuated endotoxin-induced histopathologic severity of lung parenchyma associated with neutrophils infiltration, but not neutrophil counts in BALF from rabbits. This result is consistent with the previous observation that direct insult to the lung parenchyma (intranasal instillation of LPS) increased significantly granulocyte migration in the BALF, but not indirect insult (intravenous administration of LPS) [28, 29].
In conclusion, UTI reduced LPS-induced histopathologic alteration in lung parenchyma, and it is suggested that UTI may be effective in attenuating early ALI and preventing progression of ARDS due to sepsis in the clinical setting.
References
Ware, L.B., and M.A. Matthay. 2000. The acute respiratory distress syndrome. The New England Journal of Medicine 342: 1334–1349.
Knaus, W.A., X. Sun, R.B. Hakim, and D.P. Wagner. 1994. Evaluation of definitions for adult respiratory distress syndrome. American Journal of Respiratory and Critical Care Medicine 150: 311–317.
Repine, J.E. 1992. Scientific perspective on adult respiratory distress syndrome. Lancet 339: 466–469.
Schnaitman, C.A., and J.D. Klena. 1993. Genetics of lipopolysaccharide biosynthesis in enteric bacteria. Microbiological Reviews 57: 655–682.
Suffredini, A.F., R.E. Fromm, M.M. Parker, M. Brenner, J.A. Kovacs, R.A. Wesley, and J.E. Parrillo. 1989. The cardiovascular response of normal humans to the administration of endotoxin. The New England Journal of Medicine 321: 280–287.
Martich, G.D., R.L. Danner, M. Ceska, and A.F. Suffredini. 1991. Detection of interleukin 8 and tumor necrosis factor in normal humans after intravenous endotoxin: The effect of antiinflammatory agents. The Journal of Experimental Medicine 173: 1021–1024.
Miller, E.J., A.B. Cohen, S. Nagao, D. Griffith, R.J. Maunder, T.R. Martin, J.P. Weiner-Kronish, M. Sticherling, E. Christophers, and M.A. Matthay. 1992. Elevated levels of NAP-1/interleukin-8 are present in the airspaces of patients with the adult respiratory distress syndrome and are associated with increased mortality. The American Review of Respiratory Disease 146: 427–432.
Donnelly, S.C., R.M. Strieter, S.L. Kunkel, A. Walz, C.R. Robertson, D.C. Carter, I.S. Grant, A.J. Pollok, and C. Haslett. 1993. Interleukin-8 and development of adult respiratory distress syndrome in at-risk patient groups. Lancet 341: 643–647.
Reinhart, K., and F.M. Brunkhorst. 2006. Markers for sepsis diagnosis: What is useful? Critical Care Clinics 22: 503–519.
Sato, H., S. Kajikawa, S. Kuroda, Y. Horisawa, N. Nakamura, N. Kaga, C. Kakinuma, K. Kato, H. Morishita, H. Niwa, and J. Miyazaki. 2001. Impaired fertility in female mice lacking urinary trypsin inhibitor. Biochemical and Biophysical Research Communications 281: 1154–1160.
Fries, E., and A.M. Blom. 2000. Bikunin–not just a plasma proteinase inhibitor. The International Journal of Biochemistry & Cell Biology 32: 125–137.
Tsujino, T., T. Kawabe, and M. Omata. 2007. Antiproteases in preventing post-ERCP acute pancreatitis. Journal of Orthodontic Practice 8: 509–517.
Nakamura, H., S. Abe, Y. Shibata, M. Sata, S. Kato, H. Saito, T. Hino, H. Takahashi, and H. Tomoike. 1997. Inhibition of neutrophil elastase- induced interleukin-8 gene expression by urinary trypsin inhibitor in human bronchial epithelial cells. International Archives of Allergy and Immunology 112: 157–162.
Aosasa, S., S. Ono, H. Mochizuki, H. Tsujimoto, C. Ueno, and A. Matsumoto. 2001. Mechanism of the inhibitory effect of protease inhibitor on tumor necrosis factor alpha production of monocytes. Shock 15: 101–105.
Aibiki, M., and J.A. Cook. 1997. Ulinastatin, a human trypsin inhibitor, inhibits endotoxin-induced thromboxane B2 production in human monocytes. Critical Care Medicine 25: 430–434.
Inoue, K., H. Takano, R. Yanagisawa, M. Sakurai, A. Shimada, S. Yoshino, H. Sato, and T. Yoshikawa. 2005. Protective role of urinary trypsin inhibitor in acute lung injury induced by lipopolysaccharide. Experimental Biology and Medicine 230: 281–287.
Inoue, K., H. Takano, A. Shimada, R. Yanagisawa, M. Sakurai, S. Yoshino, H. Sato, and T. Yoshikawa. 2005. Urinary trypsin inhibitor protects against systemic inflammation induced by lipopolysaccharide. Molecular Pharmacology 67: 673–680.
Sakashita, A., Y. Nishimura, T. Nishiuma, K. Takenaka, K. Kobayashi, Y. Kotani, and M. Yokoyama. 2007. Neutrophil elastase inhibitor (sivelestat) attenuates subsequent ventilator-induced lung injury in mice. European Journal of Pharmacology 571: 62–71.
Matsukawa, A., T. Yoshimura, K. Miyamoto, S. Ohkawara, and M. Yoshinaga. 1997. Analysis of the inflammatory cytokine network among TNF alpha, IL-1 beta, IL-1 receptor antagonist, and IL-8 in LPS-induced rabbit arthritis. Laboratory Investigation 76: 629–638.
Eaton, S., and G. Martin. 2002. Clinical developments for treating ARDS. Expert Opinion on Investigational Drugs 11: 37–48.
Endo, S., K. Inada, H. Yamashita, T. Takakuwa, H. Nakae, Y. Yamada, T. Kasai, K. Taki, and M. Yoshida. 1993. The inhibitory actions of protease inhibitors on the production of polymorphonuclear leukocyte elastase and interleukin 8. Research Communications in Chemical Pathology and Pharmacology 82: 27–34.
Chen, H.C., H.C. Lin, C.Y. Liu, C.H. Wang, T. Hwang, T.T. Huang, C.H. Lin, and H.P. Kuo. 2004. Neutrophil elastase induces IL-8 synthesis by lung epithelial cells via the mitogen-activated protein kinase pathway. Journal of Biomedical Science 11: 49–58.
Kuwahara, I., E.P. Lillehoj, W. Lu, I.S. Singh, Y. Isohama, T. Miyata, and K.C. Kim. 2006. Neutrophil elastase induces IL-8 gene transcription and protein release through p38/NF-{kappa}B activation via EGFR transactivation in a lung epithelial cell line. American Journal of Physiology. Lung Cellular and Molecular Physiology 291: L407–L416.
Okumura, Y., H. Inoue, Y. Fujiyama, and T. Bamba. 1995. Effects of serine protease inhibitors on accumulation of polymorphonuclear leukocytes in the lung induced by acute pancreatitis in rats. Journal of Gastroenterology 30: 379–386.
Nakatani, K., S. Takeshita, H. Tsujimoto, Y. Kawamura, and I. Sekine. 2001. Inhibitory effect of serine protease inhibitors on neutrophil-mediated endothelial cell injury. Journal of Leukocyte Biology 69: 241–247.
Owen, C.A., and E.J. Campbell. 1999. The cell biology of leukocyte-mediated proteolysis. Journal of Leukocyte Biology 65: 137–150.
Pelosi, P., D. D’Onofrio, D. Chiumello, S. Paolo, G. Chiara, V.L. Capelozzi, C.S. Barbas, M. Chiaranda, and L. Gattinoni. 2003. Pulmonary and extrapulmonary acute respiratory distress syndrome are different. The European Respiratory Journal. Supplement 42: 48s–56s.
Szarka, R.J., N. Wang, L. Gordon, P.N. Nation, and R.H. Smith. 1997. A murine model of pulmonary damage induced by lipopolysaccharide via intranasal instillation. Journal of Immunological Methods 202: 49–57.
Menezes, S.L., P.T. Bozza, H.C. Neto, A.P. Laranjeira, E.M. Negri, V.L. Capelozzi, W.A. Zin, and P.R. Rocco. 2005. Pulmonary and extrapulmonary acute lung injury: Inflammatory and ultrastructural analyses. Journal of Applied Physiology 98: 1777–1783.
Author information
Authors and Affiliations
Corresponding author
Additional information
Hong-Beom Bae and Cheol-Won Jeong equally contributed to this article.
This study was supported by a grant (CNUHRICM-Y-2007011) from Chonnam National University Hospital Research Institute of Clinical Medicine.
Rights and permissions
About this article
Cite this article
Bae, HB., Jeong, CW., Li, M. et al. Effects of Urinary Trypsin Inhibitor on Lipopolysaccharide-Induced Acute Lung Injury in Rabbits. Inflammation 35, 176–182 (2012). https://doi.org/10.1007/s10753-011-9303-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-011-9303-y